
HTT Structure & Function MFA
SUMMARY
Subsequent to the cloning of the HTT gene encoding a pioneer protein sequence, the cellular function of the protein and the dysregulation caused by the CAG and polyglutamine repeat expansions remain areas of vital inquiry. Intervention strategies that can block the gain of pathogenic function or mitigate the loss of normal function actuated by the mutant huntingtin entities (DNA, RNA, protein) provide an attractive avenue to develop huntingtin-based therapies for HD. The HTT Structure/Function MFA aims to identify the most proximal molecular mechanisms—cis modifications on the HTT gene and its encoded RNA and protein products—that can prevent the functional pathogenic consequences of mutant huntingtin and restore a normal state.
RESEARCH VISION
The pathogenic effects of the CAG/polyglutamine repeat expansion in mutant huntingtin are poorly understood, and the extent to which these effects reflect toxic gain-of-function versus loss-of-function mechanisms remain largely unresolved. Although a single gene mutation, any of the toxic huntingtin entities (gene, RNA, protein) bearing expanded repeat mutant structures could be the drivers of pathogenicity. The primary goal of the HTT Structure/Function MFA is to delineate the molecular mechanisms directly linked to mutant huntingtin that underlie HD. In a set of activities complementary to the HTT Lowering MFA, our approach investigates the therapeutic value of normalizing, reversing or otherwise intervening in aberrant huntingtin structure and function rather than solely focusing on lowering HTT levels—although we appreciate that these approaches, if successful, may converge. We postulate that identifying intervention points on mutant huntingtin to either restore normal or block pathogenic functions will point to therapeutic approaches to mitigate HD pathophysiology.
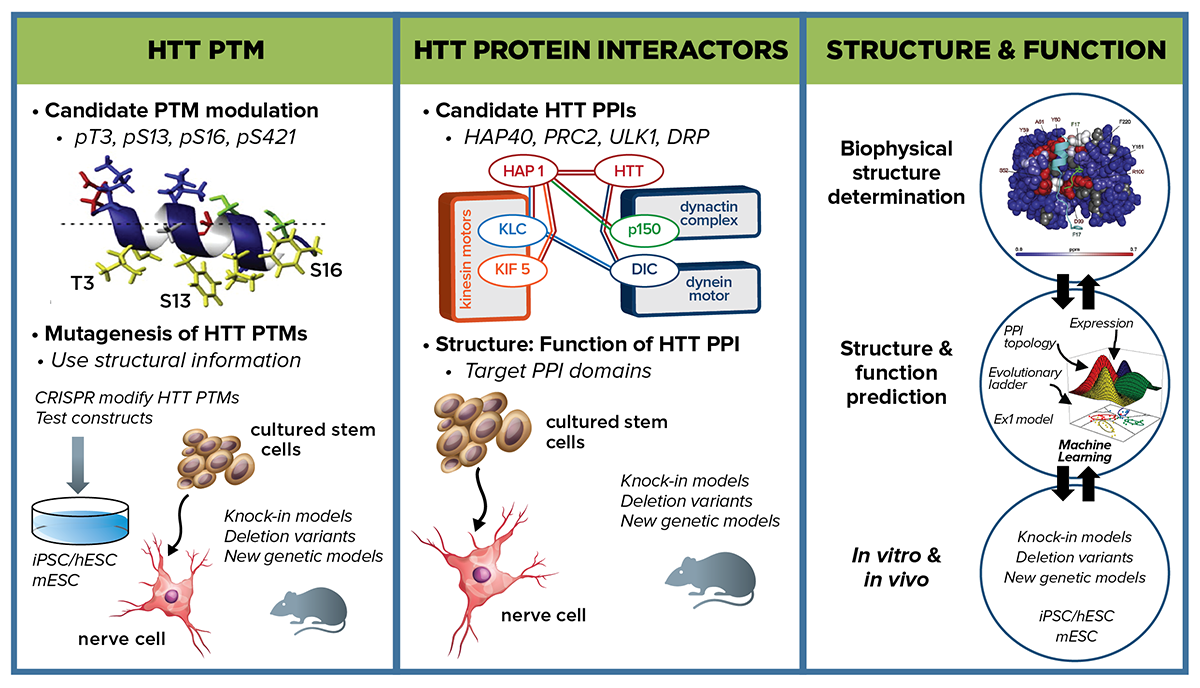
Our initial strategic priority is to focus on the HTT protein, and we will address the following critical questions: First, are there structural conformations of mHTT that can either be blocked or locked that can reduce pathogenicity? Second, can post-translational modifications (PTMs) on HTT be modulated to reduce pathogenicity? Third, are there pivotal mHTT protein-protein interactions (PPIs) that influence protein conformational states and can be targeted to reduce pathogenicity?
As a backdrop to characterizing these PTMs and PPIs, CHDI will conduct extensive biophysical manipulation and characterization of both mutant and wild-type HTT structure, as well as computational modeling of structure-function relationships. By identifying structural elements that contribute to pathology, as well as strategies to revert mHTT structure toward a normal state, we can potentially block the availability of a pathogenic PTM or inhibit an aberrant PPI, thereby mitigating toxicity.
These three areas—PTMs, PPIs, and HTT protein structure analysis—are closely interrelated. For example, some PTMs under investigation—specifically T3, and to a lesser extent S13 and S16—regulate the alpha-helical character of HTT’s N-terminal domain, and consequently its interaction with membranes and/or other protein partners integral to its function. New findings regarding PTMs and PPIs will each inform the other and guide HTT protein structure and modeling approaches. Our overall aim is to identify modulating interventions with therapeutic potential.
The pathophysiology caused by mutant huntingtin emanates from induced structural changes in not only the protein but both the mRNA and DNA too. At the nucleic acid level, one priority is to understand whether mHTT DNA and RNA CAG repeat secondary structures significantly contribute to pathogenicity. CHDI will support structural studies to identify the functional consequences of aberrant mRNA hairpins such as mis-splicing in the mHTT mRNA, mis-localization of mHTT mRNA products, and impact on regulation of HTT mRNA turnover and steady state levels. Another priority is to determine whether somatic instability of the CAG repeats in HTT DNA in HDGEC cells is either a meaningful marker or plays an important causal role in pathogenicity. We will comprehensively define both the cis-acting DNA sequences and the trans-acting factors that regulate HTT transcription to gain insight into gene regulation, mRNA production and somatic instability. Investigations in the area of DNA handling and somatic instability are increasing with accumulating GWAS evidence of DNA repair genes as modifiers. We also propose investigating whether the HTT DNA CAG expansion alters chromatin looping at the supra-level and either cis- or trans-based transcription. This fundamental knowledge of HTT mRNA and DNA is essential to understand the causal gene and will offer therapeutic insight at the DNA, RNA and protein levels.
RESEARCH PROGRAM DETAILS
Protein structure-function studies
Structure-function and biophysical modeling How would a greater knowledge of HTT protein structure inform us about both function and modulation of pathogenesis? Little is known about the three-dimensional structure of full-length HTT since, at 3144 amino acids and 350 kilodaltons, its size continues to be challenging for high-resolution biophysical techniques such as X-ray crystallography and NMR. However, details of the N-terminal structure and the mHTT structure-function relationship are now emerging. A postulate is that the root of HTT-driven dysfunction lies in a cell’s chronic exposure to aberrant mHTT protein structures, which may dysregulate PTM dynamics, promote dysfunctional interactions with protein partners/complexes, and cause wide-scale problems over time. The goal of CHDI’s structure-function investigations is to identify the most proximal interventions that act directly on HTT structure to affect function.
Strategy to identify direct intervention points on mutant huntingtin—the gene, its encoded RNA and protein products—to either restore normal or block pathogenic functions that will point to therapeutic approaches to mitigate HD pathophysiology.
With collaborators we are using a range of biophysical techniques, including X-ray crystallography and NMR, to conduct in-depth structural comparisons of wild-type and mutant HTT. We are also generating a set of purified HTT proteins of different length with various modifications and polyglutamine repeat ranges to enable this array of biophysical techniques. Manipulations of structure-function—for example, individual amino acid changes, and domain deletions like the proline-rich stretch or the N-terminal 17 amino acid (N17) region of exon 1—followed by tracking the effect on cell phenotype will allow us to further probe the function of different structural components. Such studies will help develop functional assays and guide further experiments. Examining the structural basis of the toxicity imparted by the polyglutamine expansion will identify opportunities to shift mHTT structure toward wild-type and identify pharmacological binding sites in HTT or its interactors.
Structure-function studies will also allow us to investigate several open questions: Beyond the well-studied exon 1, where are HTT’s domain boundaries and what are their molecular functions? In terms of HTT intramolecular interactions, how do they relate to or fold onto other domains? What are the properties of full-length HTT? What are the structures, interactions and functions of the nuclear localization signal, nuclear export signal, and HEAT domains, and how is each altered in the context of polyglutamine expansion?
In addition to biophysical and cellular experiments, this MFA also has a significant computational component. We are exploring a machine-learning framework to model exon 1 structure, which we later hope to expand to full-length HTT. In the longer term, we plan to combine existing high-throughput data on HTT interactions, biophysical structure and modeling data, molecular networks, and gene expression to build a model of HTT function. As an unbiased strategy using heterogeneous data and tested computational methods this would be a first-of-its-kind data-driven approach to deduce HTT function.
Post-translational modifications Our near term strategic priority is to evaluate whether specific HTT PTMs can, individually or in concert, modulate pathogenicity. CHDI and collaborators are working to produce, qualify, and make available to the wider field a range of reagents—antibodies, proteins, cell lines, tool compounds—to address this question. We plan to conduct extensive functional profiling of HTT PTMs, initially on those suggested to modulate mHTT dysfunction. We will compare HTT PTM functional readout states with respect to wild-type, CAG-expanded, and HTT null alleles. In parallel we will work to enhance and expand the range of readouts that can inform on cell-based structure-function relationships. Functional readouts will be evaluated through a suite of cellular assays reflective of HTT functions—nuclear-cytoplasmic shuttling, RNA and protein transport, lipid membrane interactions, and chromatin and transcriptional molecular signatures. More unbiased readout measures are also being developed, such as multiparametric approaches (e.g. cell painting) using a diverse set of both fixed and dynamic endpoints, which will be particularly useful for HTT PTMs predicted to affect multiple cellular mechanisms. We will also use robotic microscopy techniques that physically examine the cell at multiple time points. CHDI seeks collaborators to extend the repertoire of cell-based readouts of HTT pathogenic function. HTT PTMs that have the greatest functional effect in cell-based assays will be prioritized to evaluation in vivo in rodent HD models.
Extensive evidence supports PTMs as promising targets for therapeutic intervention in neurodegenerative diseases such as SCA1. Multiple PTMs in the N17 region of HTT exon 1 are implicated to have biological relevance to HD; for example, mutating S13 to D can rescue mHTT-associated phenotypes in the BACHD mouse model. To initiate PTM studies and develop approaches and assays, CHDI will conduct in-depth functional analyses on a select set of well-characterized PTMs—including pT3, pS13, and pS16 that have established links to HTT structure-function—to establish the PTMs state with respect to readouts of mHTT molecular pathology. For consequential PTMs, the upstream enzymatic modulators that can alter mHTT function, ideally by shifting it closer to a wild-type state, will be pursued.
Working with the Stem Cell and Regenerative Medicine MFA, isogenic allelic series of HD human and mouse embryonic stem cells with varying CAG repeat lengths, as well as HDGEC-derived induced pluripotent cells, will be used as a starting point to introduce mutations of our top-priority PTMs. Recombinase-mediated exchange ready HTT exon 1 mouse and human embryonic cell lines will facilitate a comprehensive survey of amino acid changes across the entirety of exon 1—both in individual amino acids (e.g. T3) as well as in concert (e.g. T3, S13, S16)—followed by testing in the suite of cell-based functional assay readouts. We will use CRISPR-based genome editing strategies to implement amino acid changes downstream of exon 1.
In addition to this candidate-based strategy we will also take a more unbiased approach in the search for PTMs with functional relevance to human HTT biology. HTT is decorated with dozens of different known PTMs, most of them poorly characterized. In a prioritized set of about 30 PTMs that have some rationale for functional involvement, we plan to screen in parallel across our growing suite of cell-based assays to determine whether newly-introduced HTT mutations can revert mHTT dysfunction toward a normal phenotype (or, conversely, drive wild-type HTT function toward a mutant phenotype). In both these approaches we also aim to identify kinases or other enzymes that modify PTM activity and are therefore potential pharmacological targets.
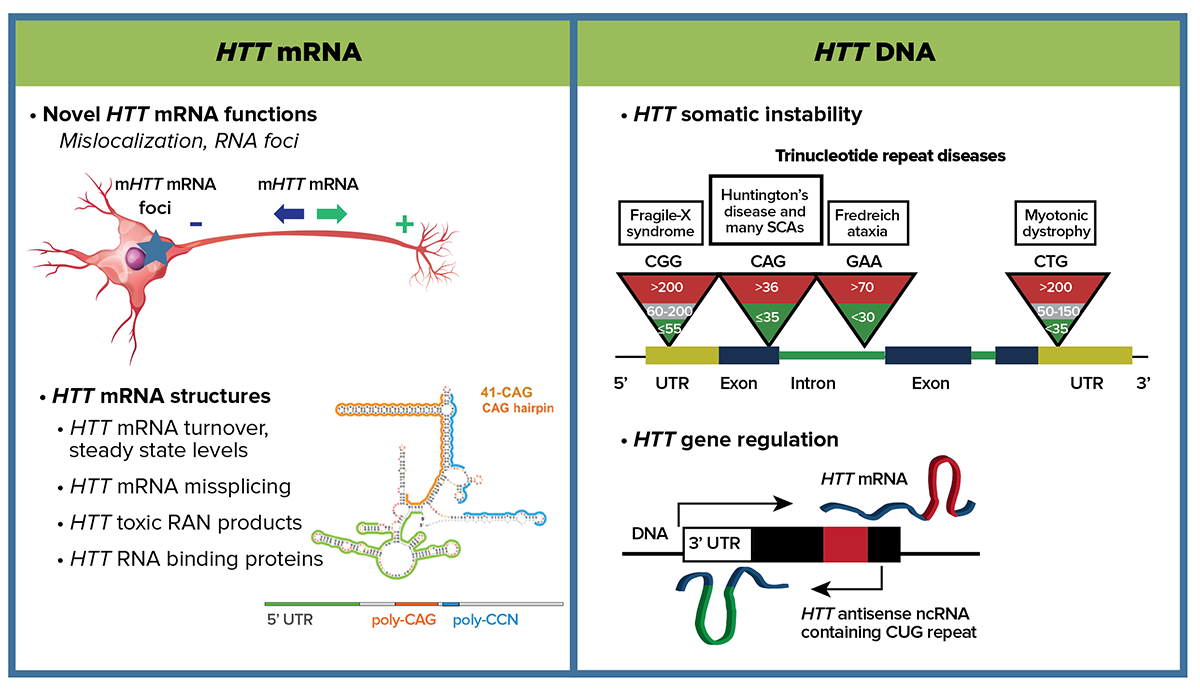
Depiction of experimental approaches for HTT mRNA and HTT DNA subareas.
Protein-protein interactions Are there changes to physiological or novel PPIs with mHTT that contribute to pathogenesis and, if so, would blocking those interactions show evidence of therapeutic benefit? We aim to pursue both prioritized candidates and unbiased approaches to investigate proteins that interact with HTT, with particular interest in proteins that preferentially interact with mHTT. We are building on existing PPI datasets to prioritize candidates, and have compiled a HTT PPI database for the HD research community, called HINT, that can be accessed at HDinHD.org. We are both revisiting prior experimental approaches with new refinements (e.g. Y2H) as well as pursuing new advances in tagging and co-IP-LC mass spectrometry approaches to identify key interactions in cells and tissues. High-priority candidates will be experimentally validated in cell and animal model systems and pursued with additional structure-function interaction mapping in wild-type and HD models. We plan to combine biochemical, genetic and pharmacological approaches to investigate the effect of disrupting HTT PPI and identify potential therapeutic targets.
RNA structure-function studies
There is secondary structure divergence within physiological CAG repeat length ranges of the HTT mRNA. The functional consequences and contribution to disease pathophysiology of this aberrant mRNA structure remain undefined. Based on evidence from other repeat expansion diseases, multimodal pathogenesis emanating from mHTT mRNA, as well as protein, may be involved in HD. We propose a focused investigation of prioritized mechanisms of mHTT mRNA toxicity that, if modulated, could have a substantial effect on HD pathophysiology.
Recent studies modeling the structure of HTT mRNA have revealed that, at pathogenic CAG repeat lengths, a highly-structured hairpin loop forms that is distinct from normal CAG length structures. We aim to target these unique secondary structures to block pathogenic consequences and/or to affect allele-specific HTT translation mechanisms. Pathogenic expanded CAG hairpin structures may provide an errant location for RNA-protein binding, which might functionally alter splicing, for example by sequestering spliceosome factors leading to an array of mis-spliced targets more pleiotropically damaging than a single alteration. Structured mRNA hairpins could alter regulation at the HTT locus, which might lead to aberrant turnover or mis-localization of the mutant transcript, mis-splicing of HTT itself, or altered translational initiations, among other scenarios. Technologies to optimize the detection of mHTT mRNA—specifically, its subcellular localization, turnover, and quantification—are also areas of high priority.
Overall, our strategy is to look to rapidly test the potential pathogenic contribution arising from altered mHTT mRNA structure. This will involve evaluating the contribution of high-priority RNA mechanisms to disease pathology, generating tools to reverse or intervene in these mechanisms therapeutically, and developing a better understanding of the biology underlying mHTT mRNA-specific regulation.
DNA structure-function studies
At the DNA level our focus is two-fold. First, we are investigating the CAG repeat somatic instability that is observed in human cells. This phenomenon is present in other trinucleotide repeat diseases; however, to date a direct pathophysiological link for HD has not been demonstrated. Our working hypothesis is that somatic expansion of CAG repeats is an important contributor to the disease process in HD and may be a modifier of pre-existing disease risk. Alternatively, we consider that somatic instability is a correlative marker of a key pathogenic process. Recent results from human GWA studies conducted within the Unbiased Systems-based Approaches MFA point to candidate gene variants that may be involved in DNA handling. The parsimonious hypothesis is that these candidate genes modify disease by modulating HTT gene somatic instability. Resolving the question of correlation versus causation is a high priority for CHDI.
Second, to address a fundamental gap in HTT gene characterization, we are also pursuing studies exploring the cis- and trans-regulatory elements and the dynamics of local chromatin structure that govern HTT expression. Comprehensively understanding regulation of the HTT gene is crucial to understanding how that process might differ in different cell types and in response to endogenous and exogenous perturbations. Characterization of the cis-acting sequences (and potential variants) as well as the trans-acting factors is relevant to potential HTT-lowering approaches operating at the level of transcription. We will also explore the potential differences in HTT chromatin structure as a consequence of CAG repeat expansion.
Suggested further reading
Saudou F & Humbert S. The biology of huntingtin. Neuron. (2016) 89:910
Vijayvargia R et al. Huntingtin’s spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function. Elife. (2016) 5:e11184
Fodale V et al. Polyglutamine- and temperature-dependent conformational rigidity in mutant huntingtin revealed by immunoassays and circular dichroism spectroscopy. PLoS One. (2014) 9:e112262
Gu X et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. (2009) 64:828
Chiki A et al. Mutant exon1 huntingtin aggregation is regulated by T3 phosphorylation-induced structural changes and crosstalk between T3 phosphorylation and acetylation at K6. Angew Chem Int Ed Engl. (2017) 56:5202
Kratter IH et al. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J Clin Invest. (2016) 126:3585
Lasagna-Reeves CA et al. Reduction of Nuak1 decreases tau and reverses phenotypes in a tauopathy mouse model. Neuron. (2016) 92:407
Ross CA & Truant R. DNA repair: A unifying mechanism in neurodegeneration. Nature. (2017) 541:34
DiGiovanni LF et al. Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum Mol Genet. (2016) 25:3937
Busan S & Weeks KM. Role of context in RNA structure: flanking sequences reconfigure CAG motif folding in huntingtin exon 1 transcripts. Biochemistry. (2013) 52:8219