
Stem Cells & Regenerative Medicine MFA
SUMMARY
The Stem Cells & Regenerative Medicine MFA will address three main questions: First, to what degree can HD pathogenesis be modeled using both human pluripotent stem cells and their differentiated derivatives in culture? Second, is HD a neurodevelopmental disease? Third, can we repair an HD brain through cell-based transplants, induction of endogenous neurogenesis, lineage conversion of non-vulnerable cells, or a combination thereof?
That HD is driven by a single causal mutation makes it especially amenable to cell-based modeling. Stem cell technologies combined with genome editing now make it possible to create pluripotent cell lines (embryonic and induced pluripotent stem cells) and their differentiated progeny as HD models. These human cell models can be characterized and compared in numerous ways to investigate the cellular basis of pathogenesis. CHDI’s overall strategy within the Stem Cell and Regenerative Medicine MFA is to create a series of pluripotent cell lines to serve as a platform to study the pathophysiological effects of mutant huntingtin on MSNs—considered the most vulnerable to the disease—and other neuronal and non-neuronal cell types in the brain. We are focused on understanding the role of wild-type and mutant huntingtin during embryonic development, as characterized in mouse and human models, with two objectives. First, understanding the signals and cellular pathways that underlie striatal development in vivo will enable better methods to differentiate stem cells in culture. Second, this information will provide a more-detailed framework to address the role of mutant huntingtin during neurodevelopment. Additionally, comparing the mutant huntingtin-related cellular signatures defined through a wide range of cell-based assays to results of ongoing studies in the mouse allelic series will provide insight into whether the gene networks identified in HD mouse studies are preserved in humans.
RESEARCH VISION
A major constraint on HD research has been the scarcity of available brain tissue from HDGECs. Advances in stem cell biology over the past 15 years have radically altered the research landscape for HD and other neurodegenerative diseases by enabling the generation of human neurons and other brain cells with specific disease properties. CHDI is working with colleagues to employ a range of techniques in genetic engineering and stem cell biology to create a diverse collection of human and mouse cell-based disease models. This collection of pluripotent stem cell (PSC) lines will include genetically-engineered human and mouse embryonic stem cells (ESCs) as well as patient-derived induced pluripotent stem cells (iPSCs).
The primary goal of this MFA is to establish a robust, carefully-validated set of PSC tools that can be used to examine both wild-type and mutant huntingtin function, as well as the molecular mechanisms underlying HD pathology in cells of the CNS, with a priority on neurons. The first set of these cell lines were generated by colleagues using a human embryonic stem cell line, RUES2, as a starting point. These cells will be thoroughly validated and standardized, and then characterized using a wide range of phenotypic, omics, and other assays.
These cell lines will serve as a research platform for CHDI that will have applications in many of the other MFAs and as an HD research community resource. For example, the RUES2 human allelic series will have increasingly long CAG repeats in one HTT allele that parallels the mouse CAG-dependent allelic series developed within the Unbiased Systems-based Approaches MFA. Working with the HTT Structure/Function MFA, we will generate mouse and human cell lines that vary in key sites of HTT post-translational modification (PTM) or in candidate modifier genes thought to contribute to HD progression.
CHDI is also exploring the possibility of using neuronal and glial precursors derived from healthy stem cells as a therapeutic strategy directed at augmenting vulnerable cell populations in the brains of HDGECs.
RESEARCH PROGRAM DETAILS
Stem cells as tools
The core of CHDI’s stem cell platform will consist of a suite of human PSC lines carrying a range of CAG repeat lengths between 30 and 70 that encompasses a range of phenotypes, including partially penetrant, adult and juvenile onset. In this allelic series, CAG repeats of increasing length will be engineered into one allele in human embryonic stem (hES) cells with an isogenic background. The same parental hES line will be used to generate cell lines that are either hemizygous for HTT or homozygous HTT null, to help interpret gain- and loss-of-function phenotypes. Due to the wild-type HTT CAG length in the parental cell line, these cells will be devoid of historical effects that accumulate with aging and disease progression, enabling the investigation of the functional effects of CAG expansion in isolation. An allelic series will also be created from human HD PSC in which CAG repeat lengths have been reduced to the healthy range to examine the effect of cellular changes that are not resolved by genome editing at the HTT gene. This collection of cell lines will serve as a resource to study HD biology more generally through the detailed characterization of the HTT gene.
To date stem cell research has been challenged by inconsistency and variability among cell lines. Cell lines generated with CHDI support will undergo extensive validation testing—including karyotype, whole-genome sequencing, CAG repeat length determination, and pluripotency status/differentiation potential—to ensure the consistency of these research tools. These assays will be combined with omics measurements such as RNAseq, epigenomic, proteomic, and metabolomic surveys as a baseline for cell phenotype. Additionally, these data will serve as a primary measurement in unbiased and targeted studies of the effect of CAG repeat length and PTMs on HTT function. CHDI will establish a centralized stem-cell informatics database that will capture the datasets generated from these assays; this will enable higher-order analysis as well as data sharing, both internally and among collaborators.
MSNs, the major projection neurons of the striatum, appear to be the cells that undergo the most pronounced changes and are the most vulnerable to degeneration during disease progression. Developing cell-based assays to study these cells is therefore a priority. CHDI is involved in ongoing work to develop improved differentiation methods and standardized readouts for the measurement of cell phenotype, enabling the direct comparison of technical improvements across different labs. CHDI is also supporting the development of protocols to differentiate stem cells to other neuronal cell types, as well as astrocytes and oligodendrocytes.
Although the directed differentiation of stem cells in culture is the most common strategy to generate MSNs and other differentiated cell types, the direct conversion of differentiated cells to neurons offers benefits not realized using stem cell models. The reprogramming of somatic cells (e.g. dermal fibroblasts) to iPSCs resets many of the epigenetic signatures associated with aging and disease progression. In contrast, recently-developed methods enabling the direct conversion of fibroblasts to MSNs using microRNA and transcription factors are thought to maintain these signatures, providing a better cell-based model for HD phenotype in adults. We will compare MSNs generated using these complementary methods.
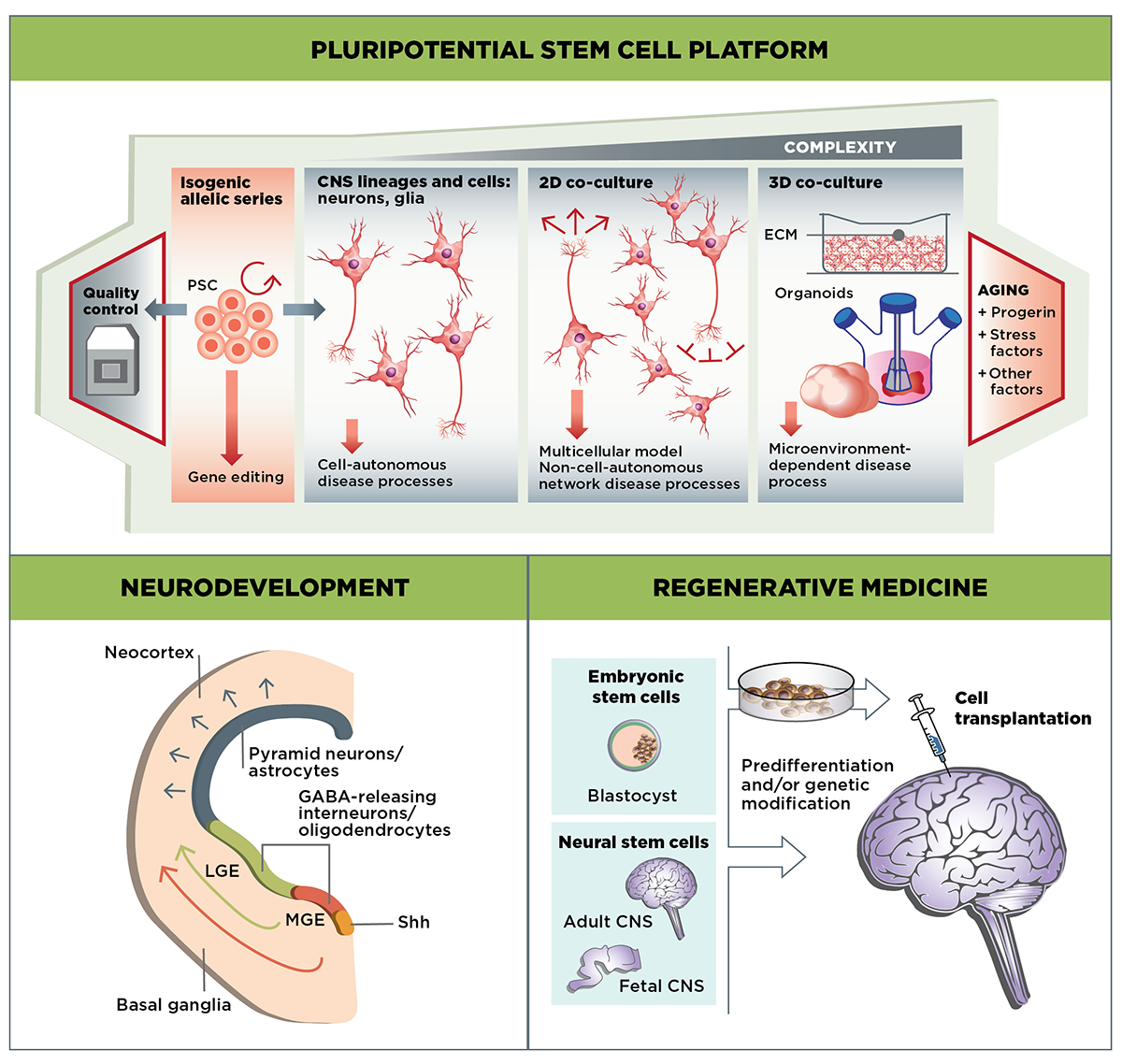
The Stem Cells & Regenerative Medicine MFA focuses on three main areas of research: Developing well-validated cell-culture HD models using stem cell-derived MSNs and other cell types; examining the role of mutant huntingtin in the developing striatum; and evaluating the potential of cell-based therapies to replace or repair the neuronal cells lost in HD.
CHDI’s intent is to create both human ESC and iPSC lines. However, we are currently prioritizing the use of genetically-engineered ESCs, which are more amenable to standardization and robust comparison between cell lines. The generation of well-characterized iPSCs from individual HDGECs also remains a priority because they offer the additional benefit of associated clinical history, a critical resource to link cellular in vitro phenotypes to HD pathophysiology and genotypic diversity. We therefore advocate collecting and banking primary peripheral blood monocytes from HDGECs, with appropriate informed consent addressed at time of collection. These cells can later be reprogrammed according to the requirements of a particular experiment, reducing cost and ensuring that each experiment uses cells reprogrammed with the same technology.
The stem cell platform will also be an important asset in understanding huntingtin biology across other MFAs. Huntingtin is thought to mediate numerous functions in cells of the CNS; CHDI and colleagues are developing a set of measurements and manipulations to define specific cellular signatures of HD that relate both CAG expansion and other structural HTT gene modifications to cellular phenotypes. These assays, based on both fixed endpoint and dynamic (live-cell) measurements, will allow us to analyze a wide range of cellular pathways that are thought to contribute to HD pathophysiology, including mitochondrial and axonal morphology and transport, HTT levels, nuclear transport, HTT localization and conformation, protein aggregation, and half-life.
In addition to experiments using isolated cell types such as neurons and glia in classical culture systems, newer assays using mixed cell populations in 2D or 3D co-culture, or in organoids, provide more accurate models to test huntingtin function in a complex cellular context. By understanding cellular phenotype through the measurement of multiple assay parameters, we plan to identify key targets and pathways that drive HD progression. Ultimately we envision that these multiparametric assays will provide critical information for HD therapeutic development.
Stem cells as targets
The range of cell-based assays, optimized for use with stem cell derivatives, will enable us to tackle key questions in HD biology that are difficult to address through other research models. First, is HD a neurodevelopmental disease? The ability to recapitulate neuronal development in culture will provide a powerful tool to examine the effect of mutant huntingtin on this process. Second, to what degree is the pathophysiology of HD a cell-autonomous phenomenon? The engraftment of stem cell-derived neurons and glia into the brains of both wild-type and HD animal models will enable us to directly examine the effect of mutant huntingtin in cells, both in isolation and in context.
Human PSC models are well suited to determining the role of mHTT in CNS development. Although HD is predominantly an adult-onset disease, this does not preclude the possibility that mutant huntingtin, which is ubiquitously present, has effects during embryonic development. Collaborators are examining the role of both wild-type and mutant huntingtin in the earliest events in neuronal development, including the generation of the three germ lineages and early events in neurogenesis. Additionally, the role of huntingtin in the development of both MSNs and cortical neurons, both of which are affected in HD, is being addressed through stem cell culture models.
Although MSNs have been a major focus of HD research, other cell types in the CNS, particularly glial (astrocytes and oligodendrocytes) and microglial cell types, may also play a critical role in disease progression. The ability to engraft glial precursor cells into the brains of HD mouse models allows CHDI collaborators to test the cell autonomy of HD. Wild-type glial precursors delay disease progression in these models, consistent with a role for these cells in mitigating neuronal excitotoxicity. These results also point to a potential avenue for cell-based therapies based on glial precursor cells.
The use of stem cell derivatives to model an adult disorder has some limitations since these cells largely reflect embryonic cellular states in vitro; characterizing the effect of epigenetic changes associated with aging on phenotypes in HD stem cell models will be critical to our understanding of HD pathophysiology in situ. The development of better stem cell models of aging remains a goal in HD research.
Stem cells as therapies
As with other neurodegenerative diseases, the use of stem cell technologies to replace or restore lost or damaged cells is an attractive therapeutic strategy in HD. A hallmark of the disease is the loss of MSNs in the striatum. Even though this loss begins well before the onset of clinical signs and symptoms, it is currently unclear whether these cells would be the best target for replacement since other cell types are now known to contribute to disease progression. For example, glutamatergic afferents provide trophic support to MSNs with the growth factor BDNF, and astroglia promote their survival through a number of possible mechanisms, including by buffering them from excitatory neurotoxicity. Glial precursor cells are currently an attractive candidate for therapeutic replacement strategies due to their ability to mitigate pathophysiology in mouse HD models. Furthermore, their wide distribution in the brain post-implantation suggests the possibility of modulating pathogenesis in the striatum and more broadly.
The timing and anatomical location for therapeutic cell engraftment also remain to be determined, and these approaches will entail regulatory issues regarding the production and safety of cell-based products.
The stimulation of endogenous brain cells to replace those lost during HD progression should also be considered, including stimulating neurogenesis in endogenous CNS stem cell populations and investigating the conversion of other differentiated cell types, such as glial cells, to assume a neuronal phenotype. Although these endogenous cells will possess mutant huntingtin it is possible that their cellular “clock” would be reset so that they function as an extended replacement for lost cells.
CHDI envisions that the treatment of HD will ultimately be comprised of a complementary set of therapies that might include disease-modifying therapies such as HTT lowering paired with therapies like cell-based replacements that can restore or repair compromised function.
Suggested further reading
Inoue HN et al. iPS cells: a game changer for future medicine. EMBO J (2014) 33:409
Onorati MV et al. Molecular and functional definition of the developing human striatum. Nat Neurosci. (2014) 17:1804
Victor MB et al. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron. (2014) 84:311
Warmflash AB et al. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat Methods. (2014) 11:847
Haston KM & Finkbeiner S. Clinical trials in a dish: The potential of pluripotent stem cells to develop therapies for neurodegenerative diseases. Annu Rev Pharmacol Toxicol. (2016) 56:489
Lo Sardo V et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci. (2012) 15:713
Benraiss AS et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat Commun. (2016) 7:11758