
Energetics & Metabolism MFA*
* This MFA has been deprioritized until such time as the requisite compelling human data described herein comes to light.
SUMMARY
There have been several long-standing hypotheses in HD research proposing a causal link between dysregulated energetics and metabolism in HD pathogenesis. Evidence to date has been contradictory and incomplete. CHDI’s first priority in the Energetics and Metabolism MFA is to conduct studies to determine whether these processes are disturbed in HDGECs and, if so, whether this disturbance correlates with disease progression. If this basic causative connection cannot be experimentally shown, CHDI will deprioritize activities and minimize resources for this research area. We are conducting a small number of focused studies that aim to identify changes in energetics and metabolism in HDGECs and relevant HD animal models. If an HD signature for energetic dysfunction is identified, we will pursue follow-up studies exploring therapeutic avenues in this area.
RESEARCH VISION
A large body of literature suggests that energetics and mitochondrial metabolism are disturbed in HD. Dramatic weight loss that cannot be accounted for by choreic movements or malnutrition has long been recognized in some symptomatic HD patients, suggesting mitochondrial uncoupling or another energetic problem. Proton nuclear magnetic resonance spectroscopy has revealed elevated lactate levels in the brains of symptomatic HD patients indicative of a deficit in oxidative phosphorylation. A CAG repeat length-dependent reduction in phosphocreatine/creatine, ATP/phosphocreatine, and ATP/ADP ratios in peripheral tissues has also been reported. Furthermore, evidence of a marked increase in oxidative stress and its resulting damaged products has been described in manifest HD patients and animal models.
However, much of the work has been inconsistent, hard to reproduce, or conducted in HD animal models with unknown predictive value. Human data has largely been collected using either peripheral or postmortem tissues, with little evidence from the living human brain. Furthermore, two of the largest interventional clinical trials in HD—which evaluated the therapeutic benefit of two nutritional supplements, coenzyme Q10 and creatine, with purported mechanisms of action related to energetics—did not meet their efficacy endpoints and provided no additional insights into the validity of the energetic mechanistic hypothesis.
Classic mitochondriopathies, such as Leigh’s syndrome or MELAS, typically involve a dramatic or complete disruption of one or more essential energetic components and therefore have very pronounced and early phenotypes. While HD shares some features with these disorders, such as the differential vulnerability of the basal ganglia, the effects observed in HDGECs are much more subtle and occur over a much longer time period. It is unclear how mutant huntingtin exerts its effects on these systems either directly (such as through interaction with mitochondrial components) or indirectly (for example by precipitating a need for increased levels of energy production). Therefore CHDI’s top priority will be to identify and characterize the precise molecular mechanism(s) responsible for any energetic dysfunction in HD.
As a framework for our strategy, CHDI has proposed three testable postulates about the role of energetics in HD. First, the presence of mHTT protein in cells results in an increased energetic demand via an as-yet unknown mechanism. Second, early in disease course cells are able to cope with this increased demand by augmenting mitochondrial output, but over time this persistent level of energetic stress results in chronic exposure to reactive oxygen species (ROS). Third, the combination of ROS and increased energetic demand eventually compromises mitochondrial physiology, redox defense systems, and results in insufficient levels of energetic output. If correct, this trajectory might explain why some major HD clinical signs often appear later in life. And because neurons have higher energy demands than other cell types, the consequences of such a dysregulation would be especially evident in neurons.
CHDI is working with colleagues on pivotal studies to test these postulates by investigating three biological mechanisms that would likely play a role in all of them—toxicity mediated by elevated ROS levels, energy homeostasis within the cell, and mitochondrial physiology. We will strive to employ methods and technologies that will avoid the experimental confounds that have too often hampered the field, and optimize the sensitivity of well-validated assays to detect sometimes marginal signals. We will continue to use genetic, pharmacological and clinical tools to better understand the precise nature of the putative energetic dysfunction in HD.
While many aspects of energetics are well conserved across species, it remains unclear how useful current preclinical genetic models of HD will be in either elucidating the nature of a putative huntingtin-driven energetic dysfunction or predicting the efficacy of candidate interventions. Therefore, whenever possible we will focus our experimental studies in humans.
Our intent is to successfully complete our experimental schema and generate a sufficiently compelling corpus of evidence to expand our approach to a larger, rational and mechanism-based Energetics and Metabolism MFA at CHDI. That will require a clear, specific, central signal early in the disease course indicating that energetic dysfunction plays a fundamental role in HD pathophysiology.
Our goal is to generate detailed evidence that identifies rational therapeutic approaches for HD within the field of energetic dysfunction. We will then model the dysfunction in HD rodents, intervene genetically or pharmacologically to correct aberrant phenotypes, and then proceed towards clinical development. Ultimately, however, we still won’t know whether energetic deficits are causal in HD until we have a therapeutic that does as intended in humans and we see the course of their disease has been successfully modified.
RESEARCH PROGRAM DETAILS
Energy homeostasis
The most prominent metabolic alterations in pre-symptomatic and symptomatic HDGECs are weight loss, region-specific alterations in cerebral glucose use and lactate levels, and aberrant activities of mitochondrial enzymes involved in glucose metabolism. Preclinical studies in HD mouse models also point to compromised metabolic regulation. What is less clear, however, is exactly how such an impairment might affect neurons in individuals with HD.
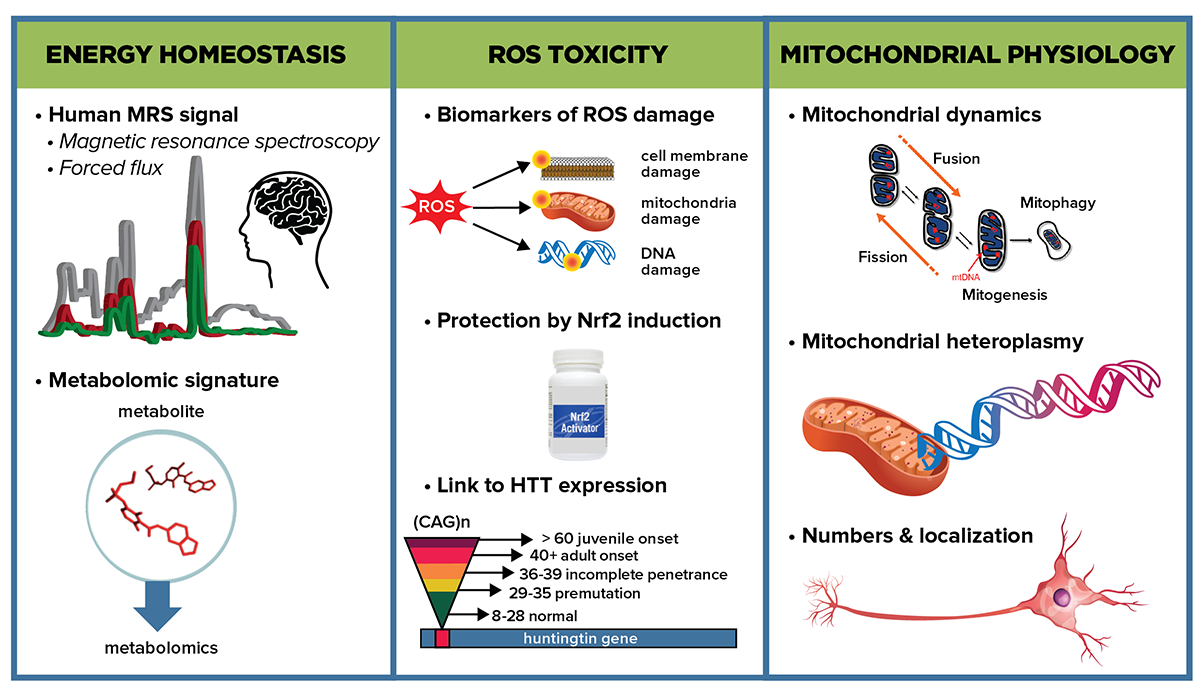
Graphical abstract depicting some of the key subareas that the Energetics and Metabolism MFA will investigate to determine energetic impairment in HD and approaches to explore the mechanisms responsible.
Although it was long thought that mitochondria do not play a role in neuronal signaling, it’s now known that a central function of mitochondrial oxidation in neurons is to supply energy for neuronal transmission. Impairment in mitochondrial energy metabolism can therefore be expected to directly affect neuronal transmission.
Recent work supported by CHDI used MRS to measure the in vivo metabolic rate in neurons and glia in two HD mouse models. The technique involves labeling glucose with carbon-13 and following that label through the tricarboxylic acid cycle to track mitochondrial energy metabolism in a chosen voxel of the brain scan. 13C MRS is especially attractive because it is completely translatable between mice and humans.
In the MRS mouse studies, both the R6/2 and Q175 HD models have shown significant reductions in neuronal energy metabolism in the striatum, as well as cortex and thalamus, before behavioral phenotypes emerged; these reductions became pronounced as disease phenotype progressed. No such reductions were observed in glial cell metabolism. These results suggest that the brain energetic dysfunction is highly sensitive to the presence of mutant huntingtin even before overt phenotypes are present and, since this dysfunction is not seen in glia, that it is due to the loss of neuronal function.
Forced flux MRS is a related technique that is conducted after first increasing the energy demands on the brain to determine whether mitochondrial metabolic activity ramps up appropriately under this stress. CHDI plans to use forced flux MRS in living human HD brains to measure a variety of energetic metabolites. We will assess correlations between observed differences to disease stage, disease severity, CAG repeat length, and regional (e.g. striatum vs cortex) or cell-type (e.g. MSNs vs glia) specificity within the brain.
This study will allow us to determine whether mitochondrial energetics are important in HD, and to understand which specific elements of the mechanism are impaired, such as glycolysis, electron transport complexes, disrupted glutamate/glutamine cycle, among others. Robust changes observed within this paradigm would provide an anchor in human biology to model this aspect of the disease. Most importantly, if deficits are uncovered we will explore therapeutic possibilities and uncover potential disease biomarkers. For example, follow-up studies would explore whether HTT-lowering strategies normalize the energetic dysfunction. On the other hand, a failure to reveal mitochondrial energetic differences in HD brains using this technique would indicate futility and consequential elimination of this MFA from CHDI’s portfolio.
It’s likely, however, that the results of this human experiment will not be so easy to interpret; considering how prevalent energetic anomalies are in neurodegenerative disease, we are likely to find some differences in HD energetics whether they are causative, compensatory, or just epiphenomena. Perhaps a more realistic aspiration for this study is to identify an HD signature of metabolites that are increased or decreased.
ROS toxicity
Oxidative stress—an imbalance between ROS production and the antioxidant defense system—has been implicated in HD pathogenesis both in humans and in animal models. Independently, two groups have reported increased markers of oxidative stress damage products in plasma and blood of manifest HD patients. Concordantly, a reduction in antioxidant systems was found and these changes correlated with disease progression. However, evidence that reducing ROS can slow disease progression is still lacking and mainly precluded by a lack of sensitive and specific quantitative methods to track various types of ROS damage. In addition, a number of confounding factors such as smoking, age, sex, diet, and sample collection procedures profoundly influence ROS measurements.
CHDI is developing and validating sensitive mass spectrometry-based methods to measure the molecular products of the deleterious effects caused by ROS to proteins, lipids and DNA. Using these ROS-byproduct assays, CHDI and colleagues will investigate increased ROS damage in HDGECs and in animal models of HD. This quantitative in vivo panel of oxidative stress markers can then be correlated with neurodegeneration and phenotype progression in both animal models and HDGECs. Once established and validated these ROS-byproduct assays will be made widely available to the HD community to evaluate oxidative stress.
To date, antioxidant therapies tested in clinical trials have shown no therapeutic benefit in any disease and it’s unlikely that any single antioxidant would be sufficient to dampen the effects of ROS in HD. If these proposed ROS marker studies identify an increase in ROS damage products in individuals with HD, the most promising therapeutic avenue would likely be coordinated increases of endogenous antioxidant defenses. CHDI plans to investigate two possible therapeutic targets along these lines: NRF2, a gene that induces innate antioxidant responses to oxidative stress, and ATM kinase, which is involved in the DNA damage response and shows elevated activity in HD.
We are developing brain-penetrant NRF2 activators and ATM inhibitors and will evaluate their effect on the oxidative stress marker panel, forced flux MRS, behavior, and other outcome measures. Another avenue of investigation is whether other pharmacological interventions, such as those that lower mutant or total HTT levels, can decrease ROS damage as measured by the oxidative stress marker panel.
We are also working with collaborators to develop pharmacodynamic markers for NRF2 activation using reporter mice as a prerequisite to assessing therapeutic potential. Using both NRF2-transgenic mice and viral delivery of NRF2, these collaborators are addressing whether NRF2 overexpression in neurons or glia can ameliorate phenotype.
Mitochondrial physiology
Mitochondrial dynamics—processes such as mitochondrial fusion and fission, trafficking, biogenesis and mitophagy—are crucial for the organelle’s function and homeostasis. Mitochondrial fragmentation and increased apoptosis has been reported in the presence of mutant huntingtin in neurons in both cell and animal models of HD, providing evidence for a pathological link between mutant huntingtin and abnormal mitochondrial dynamics. Cumulative mutations in mitochondrial DNA might also be involved in disease pathology.
CHDI is working with colleagues to further characterize mitochondrial dynamics and metabolic signatures in HD model brain tissue and various HDGEC peripheral tissues from the EHDN Multi-Tissue Monitoring (MTM) project. We are also working with other collaborators to examine the role of mitochondrial fusion and fission in HD. In cellular models, reduction in mitochondrial fission improves cell viability, suggesting that molecules promoting this process might have therapeutic potential. We are testing this idea in vivo using a mouse model in which Mff, the receptor of a protein central to mitochondrial fission, has been inactivated. These mice have extensive cardiomyopathy and mitochondrial dysfunction that can be rescued with the concomitant removal of a protein that plays a central role in mitochondrial fusion. These findings point to the balance between the two processes being important in both mitochondrial physiology and maintaining tissue and organ function. The next step of the project is to explore the role of Mff in an HD mouse model.
Another project is examining whether and how mutations and copy number variation in mitochondrial DNA might be involved in HD age of onset, disease progression, severity, or other pathological features by sequencing mitochondrial DNA from HDGECs and comparing them to those of participants in the 1000 Genomes Project. Early results suggest that cell lines derived from HDGECs have a significant increase in heteroplasmies—combinations of mutated and non-mutated mitochondrial DNA within any one cell.
Suggested further reading
Mochel F & Haller RG. Energy deficit in Huntington disease: why it matters. J Clin Invest. (2011) 121:493
Browne SE & Beal MF. Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal. (2006) 8:2061
Carroll JB et al. Treating the whole body in Huntington’s disease. Lancet Neurol. (2015) 14:1135
Tunez I et al. Important role of oxidative stress biomarkers in Huntington’s disease. J Med Chem. (2011) 54:5602
Johnson DA & Johnson JA. Nrf2 – a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med. (2015) 88:253
Chen H & Chan DC. Mitochondrial dynamics – fusion, fission, movement, and mitophagy – in neurodegenerative diseases. Hum Mol Genet. (2009) 18(R2):R169
Kim J et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet. (2010) 19:3919
Ye K et al. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci U S A. (2014) 111:10654