
DNA Repair & Handling MFA
SUMMARY
Human genetic studies implicate DNA repair genes in modifying the progression of Huntington’s disease (HD). Since somatic instability is a hallmark of HD, this raises the possibility that DNA repair complexes contribute to pathology. These converging lines of evidence in humans, as well as data from model organisms, have convinced CHDI that a new DNA Repair & Handling MFA is warranted. DNA repair genes play an important role in the maintenance of both nuclear and mitochondrial DNA integrity in neurons and other cell types, and are also known to influence trinucleotide repeat expansion events. Our initial goal in this MFA is to investigate whether interventions in DNA repair pathways revealed in human genetics studies have therapeutic potential.
RESEARCH VISION
There is growing evidence from genome wide association (GWA) studies in HDGECs that altered DNA metabolism is not only a consequence but also a significant contributor to HD pathogenesis. The priority for this MFA is to address the following inter-related questions: At which step in the DNA repair pathway(s) is it most effective to intervene, how best to modulate activity, how to assess the efficacy of modulators, and when in the pathophysiology of HD are the best therapeutic opportunities?
Our current collaborative research aims to further investigate DNA repair pathways using cell-free, cell-based, and in vivo studies to generate preclinical mechanistic and efficacy data to justify advancing therapeutic candidates to clinical evaluation. DNA repair proteins can influence CAG-repeat expansion in both the germline and somatic cells in a tissue and age-dependent manner. We plan to determine whether somatic instability is a primary pathogenic mechanism by which candidate causal genes identified in human genetic studies modify HD. This MFA will utilize human stem cell and HD animal models carrying an allelic series of HTT CAG sizes to study DNA repair pathways. We will focus on additional DNA repair network candidates identified in our ongoing human genetics discovery efforts conducted under our Unbiased Systems-based Approaches MFA. We will evaluate HTT-lowering tools that bind HTT DNA, modulate HTT transcription, or HTT RNA processing for their influence on DNA metabolism. We will also develop new pharmacologic agents, molecular tools, and genetically-engineered cells and animals to test the effect of modulating DNA repair gene candidates on HD model phenotypes. Our current plan is to identify therapeutic agents targeting the MutSâ protein complex, with an initial focus on MSH3, and to pursue this program as an exemplar for developing new candidate therapies from human genetic modifier findings.
RESEARCH PROGRAM DETAILS
Within this MFA we are looking to identify which genes within the DNA repair pathway affect the onset, severity or progression of HD pathophysiology. We will employ a variety of approaches, including human genetics, network reconstruction, and targeted manipulation of pathway members using molecular genetics and pharmacological tools, ensuring that we prioritize human evidence in selecting potential therapeutic candidates.
Identifying candidates for therapeutic intervention
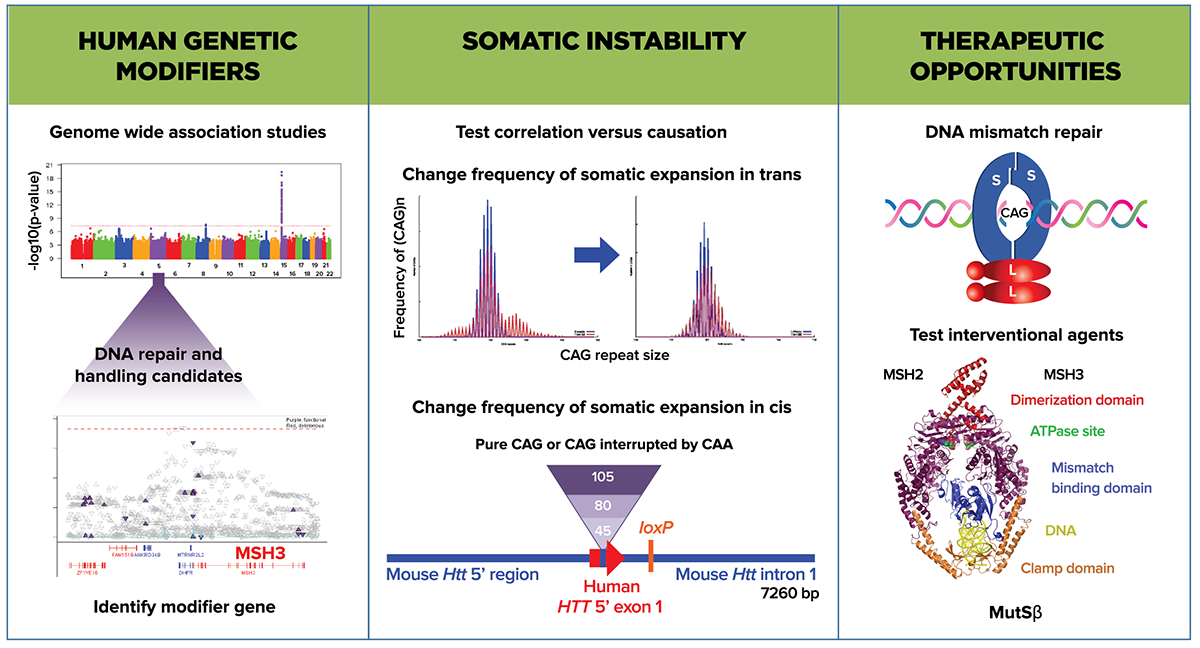
The primary driver for forming the DNA Repair & Handling MFA is the data from GWA studies implicating a number of DNA repair and genome stability pathway molecules as candidate modifiers of the age at motor onset. These findings do not explain the biochemical mechanism or indicate that there could be interventional benefit in adult HDGECs. However data from HD models does provide mechanistic insight, such as loss-of-function mutations in DNA repair genes in mouse and fruit fly HD models that alter somatic expansion, mHTT aggregate distribution, and abnormal behavioral phenotypes. mHTT itself also appears to trigger a DNA damage response, and HTT may participate in DNA repair activities. Extensive gene expression profiling in the mHTTmouse gene allelic series has described CAG length-related gene sets enriched for DNA repair and p53 signaling. The candidate genes described in these studies include proteins and complexes that mediate DNA repair via interstrand cross-link, mismatch, double-strand break, and non-homologous end-joining mechanisms. We continue to build our understanding using known networks to uncover the relationships of candidate genes identified in GWA studies and the mechanism of their effect on HD phenotypes.
We currently prioritize components of mismatch repair and genome stability identified as causal genes in GWA studies for further investigation. However, these genes may not have the largest effect on HD onset or progression, and there is a need to explore potential partners and pathway members that would have a greater influence in HD models and have greater therapeutic potential. In ongoing GWA studies that use more HDGEC samples a number of new modifier loci include additional members of DNA repair pathways. Our objective is to understand the function of these genes and how they influence disease modification.
Canonical DNA repair pathway members have been studied extensively in humans and model systems in the context of their contribution to initiating, supporting or preventing tumorigenesis. Computational biology and modeling will inform us on the role of DNA metabolism in HD and neurodegenerative diseases in general, with potential approaches such as; 1) augmenting the use of traditional bioinformatics and computational biology techniques with the existing oncology data; 2) reconstructing DNA repair pathways in the cells of the central nervous system (CNS); and 3) modeling the enzymatic reactions of DNA repair proteins to predict the effects of pharmacological intervention in those pathways. We will address the paucity of information on the role of these pathway members in the CNS, and specifically in post-mitotic neurons. A particular area of interest is to explore transcription-coupled repair and R-loops.
We plan to combine the entirety of this human genetic data (from the HD and oncology spheres) with experiments in HD model systems, such as human pluripotential stem cells and their derivatives and in mouse genetic models. Studies are underway to target candidates by knockout, knockdown, overexpression, point mutation, or chemical perturbation to test the consequences on phenotypes such as instability of the CAG repeat, mHTT aggregation, or animal behavior. We also aim to determine which cell types are the most relevant to disease modification: Are neurons the most sensitive cell type with respect to DNA metabolism’s role in HD, or is it glial cells or immune cells that are critical? Is the DNA repair pathway operating in dividing cells the same as that in post-mitotic neurons? What is the relationship between differential cell vulnerability and the modifier genes? How do the GWAS modifiers inform on triplet repeat disorders generally, and HD specifically? Supporting research to address these questions will be critical to our strategy.
Within and across these endeavors we include HTT as a candidate DNA repair pathway member and we will support investigations of its effect on DNA metabolism and trinucleotide repeat expansion.
The DNA Repair & Handling MFA is focused on identifying HD-relevant components of DNA repair pathways, understanding their mechanism of action in HD, and developing therapeutic approaches targeting these candidates.
Mechanism of action
Further insight into the GWAS-identified candidates’ mechanism of action will give us a better idea of their therapeutic potential. One hypothesis is that these candidate genes modify disease by modulating mHTT somatic instability. The lines of investigation addressing this mechanism are now within the remit of the DNA Repair & Handling MFA rather than the HTT Structure & Function MFA, and these include; (1) sequencing of the HTT CAG repeat region in humans and correlation with penetrance and disease progression; (2) longitudinal studies in HDGECs to correlate frequency and extent of somatic expansion with phenotypic delay or onset; (3) intervention to up- or downregulate DNA repair gene activity and correlate direction with HD model phenotypes; and (4) identifying interventions that block expansion and/or promote contraction of the HTT CAG repeat.
We will also examine the timing and degree of somatic instability as the phenotype manifests and progresses in humans in order to evaluate the consequence or cause of instability on HD pathophysiology. We will look for further evidence of links between somatic instability and disease onset or progression. We will also address whether somatic instability in blood cells can serve as a biomarker to optimize therapeutic interventions that target DNA repair proteins.
Similar studies looking to extend previous findings correlating somatic instability in HD animal models with pathology markers are underway, including new HD mouse models that carry glutamine-encoding CAA interruptions within the CAG repeat sequences to stabilize expansion at the HTT locus. Our aim is to provide convincing evidence that HTT CAG somatic instability and expansion are causal to HD pathology.
It is important to better understand tissue and cell-type specificity and stability of CAG repeats in different cellular contexts. We will look to measure instability in accessible tissues and build evidence to support using those measures as a predictor of CNS effects. One obstacle to this type of exploration is the stochastic and nonlinear frequency of CAG expansion events. An important gap is a method to measure HTT CAG instability in a single cell in situ or visualize different CAG lengths across cell populations in the CNS. While sensitive instability measures have been developed for use in tissue and cell populations, we will support innovative approaches to measure HTT CAG length in single cells.
Current data strongly support the link between mismatch repair biology and CAG repeat instability at the HD locus, but a direct demonstration that perturbing the pathway later in development can alter HD pathophysiology is missing. The biology is challenging to model in the mouse as CAG repeats in the 40-50 CAG range do not expand until late in its lifetime. We will use human cell-based and animal HD models to investigate how altering DNA repair protein expression or activity at different stages affects the relationship between CAG length and repeat instability. Directed studies are aimed at further destabilizing CAG repeats of largely non-phenotypic HD models to determine whether accelerated slippage of repeats in somatic cells can mimic phenotypes shown by HD models with longer inherited CAG lengths.
In HD models, components of the mismatch repair machinery are important for both intergenerational germline and somatic expansion of the CAG region. We have collaborations underway to explore intergenerational stability in HD as we look to compare the mechanism of instability in the germline to somatic instability and to onset and progression.
Our working hypothesis is that somatic expansion of CAG repeats is an important contributor to the HD disease process. Alternatively, it is possible that somatic instability is a correlative marker of a yet unidentified key pathogenic process that involves some of the mismatch repair pathway members. In both scenarios measuring somatic instability will be instructive for advancing therapeutic candidates. Resolving the question of correlation versus causation is a high priority.
Several lines of evidence describe the influence of DNA metabolism on neurodegeneration without insight into the underlying biology and mechanism of action; these include evidence for aberrant DNA damage response in Parkinson’s and Alzheimer’s disease, plus mutations identified in DNA repair enzymes that cause rare genetic disorders characterized by neurodegeneration. It is possible the activities of these proteins on global cellular processes such as chromatin organization, epigenetic modification, and gene expression also influence neurodegeneration in HD, and the DNA Repair & Handling MFA will look to study the effects of modulating DNA repair proteins on these processes.
Therapeutic opportunities
For DNA repair candidates, questions arise regarding how this type of therapeutic strategy would work and whether it is likely that selectively targeting HTT will be successful. Which DNA repair proteins or pathway or protein complex members alter CAG repeat stability and other HD phenotypes favorably when manipulated in an adult HDGEC? We plan to build a portfolio of tools to investigate components of mismatch repair (MutS and MutL, as well as other candidates such as FAN1 and RRM2B) to understand their mechanism of action and how they might interact and influence each other.
Members of the mismatch repair pathway are candidate causal modifiers, and among these human and mouse genetic data suggests reduction or loss of MSH3 activity is not as deleterious in promoting tumorigenesis as other mismatch repair components. The MutSâ complex (a heterodimer composed of MSH3 and MSH2) will be investigated as our initial exemplar for therapeutic development. MutSâ recognizes mismatches made up of loop DNA structures and then recruits a MutL homolog complex (comprised of heterodimer species involving MLH1 and various partners) and subsequently DNA repair proteins required for cleavage, destruction and repair of DNA. The MutSâ complex has well defined DNA binding, nucleotide binding, and ATP hydrolysis activities that are linked and, as such, represents a pharmacologically-tractable target. Evidence from humans and HD models suggests that inhibitors of MSH3 may have benefit in HD. Our drug-discovery approach will be to screen for enzyme inhibition, protein partner interference, and potential agents that can knock down the gene; currently, as we cannot predict which of these aspects of MSH3 activity will confer the greatest efficacy or the optimum risk/benefit profile, these will be investigated in parallel. In addition, we will generate a set of protein complex structure-function reagents, biochemical and cell-based assays, and genetically-modified animals to advance mechanistic and pharmacological studies.
As we progress we will build on the two main drivers that inspired our MFA: HD modifiers identified by human genetics and somatic instability observed in humans. Understanding the role of modifier genes in HD pathogenesis will be crucial to developing candidate therapeutics.
Suggested further reading
Dragileva E et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. (2009) 33:37
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) 162:516
Hensman Moss DJ et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. (2017) 16:701
Iyer RR et al. DNA triplet repeat expansion and mismatch repair. Annu Rev Biochem. (2015) 84:199
Kaplan S et al. A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput Biol. (2007) 3:e235
Madabhushi R et al. DNA damage and its links to neurodegeneration. Neuron. (2014) 83:266
Ross CA & Truant R. DNA repair: A unifying mechanism in neurodegeneration. Nature. (2017) 541:34
Swami M et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. (2009) 18:3039