
Neuronal Dysfunction MFA
SUMMARY
HD is characterized by extensive cell loss throughout the CNS, especially in the basal ganglia nuclei and the cortex, with the cortico-striatal-thalamo-cortical circuitry thought to be particularly affected. Recent decades have brought fundamental revisions to our understanding of basal ganglia macrocircuitry on a physiological and information processing level. However, this new understanding has not yet been adequately applied to current theories of the emanation of circuitry dysfunction in HD, or of its correlation to phenoconversion, disease progression and clinical sign and symptom presentation. CHDI’s Neuronal Dysfunction MFA aims to generate a complete conceptualization of circuitry dysfunction and to elucidate both the mechanistic link to mutant huntingtin and how it drives HD progression. This will be accomplished through in-depth comparisons of circuitry dysfunction in multiple HD models, expanding human data on cortico-striatal-thalamo-cortical circuitry and basal ganglia function, and the pursuit of therapeutic approaches to target CNS dysfunction and degeneration on multiple levels.
RESEARCH VISION
The main focus of the Neuronal Dysfunction MFA is to improve our understanding of how CNS circuitry dysfunction progresses during the course of HD, and how this progression drives HD signs and symptoms. Without this conceptualization we consider that our ability to develop therapies that are meaningful to patients will remain limited. Even therapies that aim to address the most proximal cause of the disease, by lowering mHTT itself, will require this insight given that currently contemplated modalities will likely have restricted distribution within the CNS and lowering mHTT in symptomatic patients may be insufficient to provide substantial clinical benefit.
Comprehensively delineating CNS circuitry dysfunction in HD will require synthesizing neurophysiological data from HDGECs and diverse HD model systems, and employing computational neuroscience approaches to build a cohesive human-relevant understanding of pathophysiology. We will then work to elucidate the molecular causes underlying dysfunction, which will point us towards new therapeutic opportunities.
To further identify promising therapeutic intervention points, we have five main goals. First, we strongly support investment in experimental medicine studies designed to assess the circuit etiology of HD signs and symptoms in specific behavioral domains—motor, cognitive, affective. Second, to optimize clinical to preclinical translation, we are generating and neurophysiologically characterizing novel rat and nonhuman primate (NHP) HD models that will allow both closer parallel comparison with human basal ganglia function and refinement of behavioral tasks to investigate HD-relevant symptoms. Third, as required, we will pharmacologically develop novel clinical candidate compounds against validated targets with the potential to affect circuit function and combat structural neurodegeneration in HD. Fourth, we support further investment in a molecular understanding of huntingtin’s role both at the synapse and in the control of key neuronal and astrocytic microcircuits. Fifth, we plan to further explore the nascent understanding of huntingtin’s role in oligodendrocyte maturation, as well as mutant huntingtin’s role in white matter and demyelination pathology.
RESEARCH PROGRAM DETAILS
Neurobiological substrate of symptom expression
Numerous unanswered questions about the etiology of HD can be framed in terms of circuitry. For instance, it’s unknown whether the persistent presence of mutant huntingtin is required for disease progression, and therefore whether its removal would change disease course. Such questions can be rephrased to ask at what point brain circuitry dysfunction becomes irreversible even if further mutant huntingtin expression is suppressed, and how determinant mutant huntingtin is during neurodevelopment and early adulthood.
These questions are currently difficult to answer in HDGECs. Clinical trials of therapies that aim to suppress mHTT translation (or mHTT transcription) are currently constrained by the limited spatial distribution these treatments achieve, and the slow disease progression imposes long delays between treatment onset and measurable effects. Additionally, recruiting sufficient HDGECs at different disease stages is impractical.
Model systems are therefore essential to elucidate circuitry dysfunction. However, gaining the deepest possible insight from these requires bridging the gaps between data from HD experimental medicine studies and conceptual models of circuit dysfunction, incorporating the more detailed information gleaned from circuitry dysfunction in multiple mammalian HD models. Combining human and model data into a single disease framework will allow us to characterize the pathogenic sequence of events at the circuitry and neurochemical levels and infer compensation or causality, as well as to identify both potential strategies/targets and stages at which disease mechanisms can still be reversed or altered.
Therefore a key goal is to conduct intensive longitudinal and cross-sectional (over age) comparisons in different species to correlate symptoms and behavioral domains with circuit dysfunction. Three knock-in HD rat models carrying either 60, 110, or 130 CAG repeats have recently been created, and our aim is to establish detailed circuitry-level phenotyping in one of these to complement similar investigations already underway in the Q175 knock-in mouse. Our emphasis is on establishing the neurophysiological basis of behavioral phenotypes in assays querying controlled, specific, delimited animal behaviors relevant to HD, such as reward seeking, executive function, and decision making. We will do this by utilizing concurrent electrophysiological, optogenetic and neurochemical in vivo methods to characterize the identified circuit dysfunction and its relationship to behavioral phenotype.
Another major priority is developing and validating NHP HD models. The relative similarity of the complex neuroanatomy of cortical and subcortical circuits in NHPs and humans, as well as the equivalence of behavioral tasks, will be invaluable. These NHP HD models will also help bridge findings between rodents and humans and, over the long-term, CHDI is working with colleagues to develop both transgenic and CRISPR/Cas9-engineered knock-in NHP models. More immediately, CHDI will aid in the neurophysiological and behavioral assessment of an existing NHP HD model (engineered via virally-delivered mHTT fragment cDNA to the caudate putamen) and characterize disease progression through imaging studies. In preparation, CHDI and colleagues are developing a suite of relevant behavioral tasks to deploy in NHP HD models as they become available; these are currently being validated in non-HD NHPs.
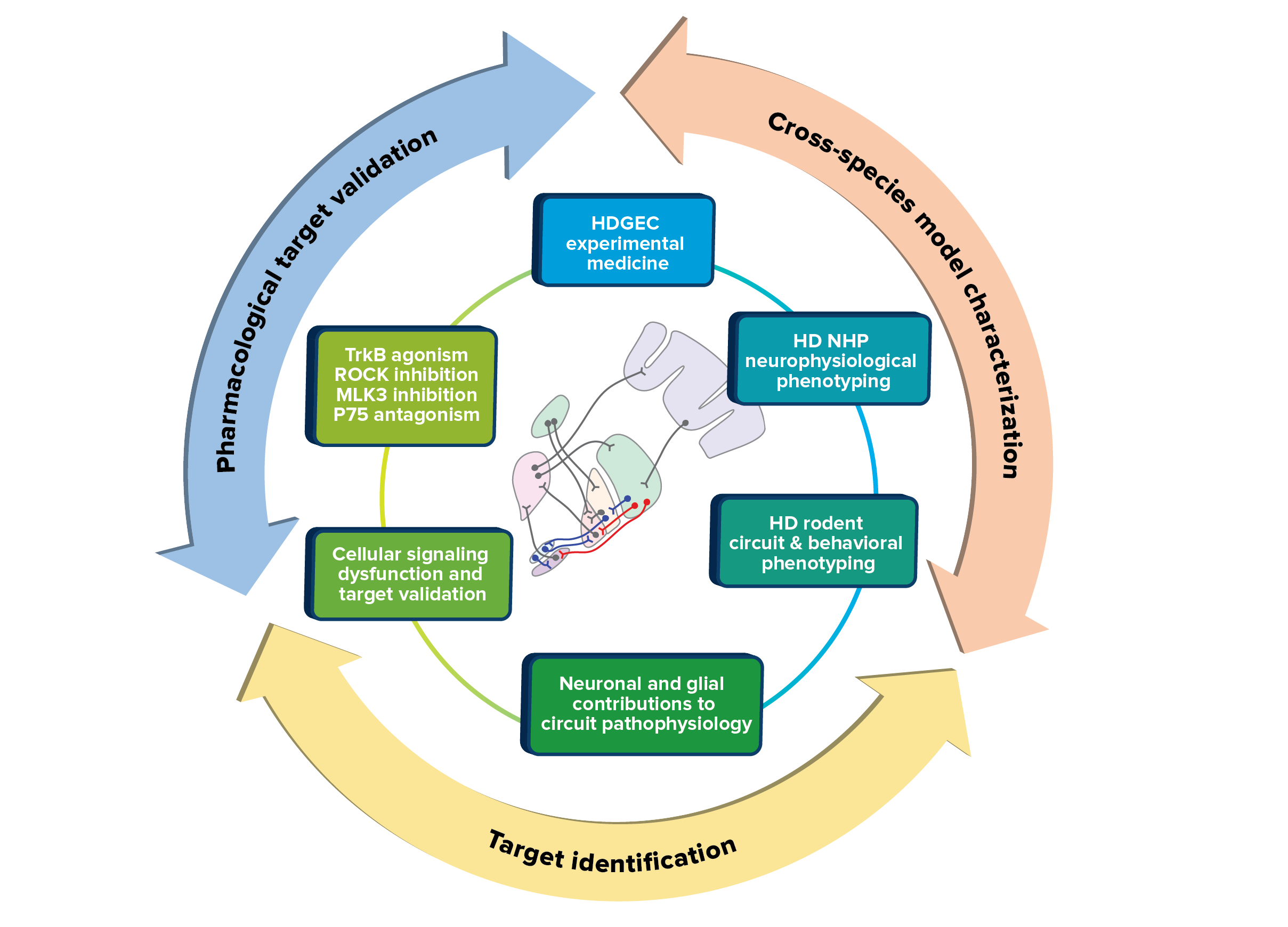
Depiction of the approaches that the Neuronal Dysfunction MFA are using to optimize translational development of HD therapeutics.
Ultimately, bridging the translational gap in our understanding of findings from HD animal models will require a more aggressive and systematic gathering of clinical data. Studies in HDGECs have chronicled marked white matter loss, cortical mantle thinning, reduced functional cortico-cortical and cortico-striatal connectivity, subcortical changes, and disrupted oscillatory patterns preceding or concurrent with cognitive, motor, and affective dysfunction in HD. The degraded connection between the cortex and basal ganglia, in combination with the disrupted information flow within the basal ganglia and between cortical areas, is likely the neural basis of many HD phenotypes. We consider that focused investigations in these areas will reveal HD-relevant network or circuit alterations that may indicate points of intervention.
A pressing need is to establish insightful correlations between assessments at similar levels of system complexity from humans and animal models. We will further encourage experimental medicine studies in HDGECs—through the Enroll-HD clinical platform and independently—to characterize, with greater precision, the early motor, cognitive, psychiatric, and affective symptomatology and its progression, in correlation with imaging and electrophysiological techniques. For example, directly measuring basal ganglia function through recordings from pallidal neurons obtained peri- and post-operatively during deep brain stimulation procedures offers a unique opportunity to gather such data in HD patients. Similarly, we aim to establish correlates of disease progression utilizing data from resting state network/default mode network in HD patients, imaging (MRI, DTI, ASL, PET), and functional assessments (fMRI, qEEG, MEG) in HDGECs, for which preclinical equivalents can be obtained from rodent and NHP HD models.
Ultimately, these diverse human and HD model datasets incorporating newly-generated preclinical behavioral, electrophysiological, neurochemical, and immunochemical data will be combined to build a computational neuroscience framework that allows query-driven analyses of HD symptom etiology.
Molecular basis of basal ganglia dysfunction
Although imaging and neurophysiological techniques to investigate pathophysiology of CNS function and neurodegeneration in humans in situ has advanced substantially, we must acknowledge that there are still severe limitations to what can be achieved in HDGECs. Many of our current hypotheses about CNS circuitry dysfunction in HD are derived from neuropathological investigation of HD postmortem brains. The structural degeneration documented by this approach largely describes pathology in individuals who were symptomatic over many years. While the heterogeneous cell loss in the CNS must result in the complete removal of elements of the affected circuit (and a likely ‘rewiring’ of compensatory circuits), we assume a priori that the malfunctioning of specific neural circuits, prior to cell death, is likely to be an important contributor to HD symptom manifestation. Studying this stage of dysfunction is critical to our understanding of disease progression and is the most likely to yield rationally-designed therapeutic interventions. In practice this is only possible in animal models that mimic, as closely as possible, HD pathophysiology.
To more cohesively understand neuronal dysfunction, CHDI and colleagues will extend our ongoing pursuit of detailed circuit phenotyping in the Q175 heterozygous knock-in mouse. In close collaboration with our academic colleagues, we are expanding our knowledge of the mutant huntingtin-induced cortico-striatal-thalamo-cortical circuit dysfunction through detailed ex vivo and in vivo interrogation of the functioning of all individual nuclei and key cell types within this entire canonical circuit. Our (possibly controversial) choice to advance one specific model is imposed by practical limitations and to avoid the difficulties in collating disparate data from multiple HD mouse models into a single conceptual framework. Choosing the Q175 mouse model was predicated largely on its suitability in evaluating allele selective Htt-lowering strategies (in support of the HTT Lowering MFA approach), having mHTT/HTT construct validity, albeit with an expanded CAG repeat. Molecularly, the Q175 model shows human HD-consistent transcriptional dysregulation, and a robust differential proteomic signature, which is being mined exhaustively by the Unbiased Systems-based Approaches MFA. Also, the Q175 heterozygous model is one of the few knock-in heterozygous models that has demonstrable and exhaustively characterized behavioral signatures with respect to motoric, cognitive, and motivational phenotypes.
Extensive breeding work (for instance to Cre-reporter lines) and copious preliminary data generation have already been undertaken in the Q175 model, which aids detailed neurophysiological circuit phenotyping both in live-cell identification, optogenetic, and cell-specific perturbation experiments. Detailed circuit mapping can also be achieved in this way. This project largely reflects a natural extension of ongoing work. Knowledge gained from this approach has already identified many novel mutant huntingtin-driven neurophysiological phenotypes apparent throughout the entire basal ganglia circuit, such as in corticostriatal connections, the striatum and striatal output pathways, and in the downstream nuclei, such as the globus pallidus, substantia nigra and STN. A much more detailed understanding of intrastriatal microcircuit dysfunction is also emerging. Of particular interest, in addition to cell-autonomous neuronal dysfunction, is the emergent role of mutant huntingtin-induced astrocyte and oligodendrocyte pathophysiology that contributes to network level dysfunction in HD, an area that will be further investigated.
Such detailed phenotyping is a prerequisite to characterizing mutant huntingtin-driven neural dysfunction, but a major focus of this MFA is to move beyond phenomenological phenotyping towards a precise understanding of the molecular events causal to the characterized dysfunctions. An understanding of the causal signaling pathway disruptions, proximal to the mHTT insult, that primarily contribute to neurophysiological phenotypes is the ultimate goal for therapeutic intervention.
Assisting design of HTT-lowering clinical trials
A major asset here is the collaborative development of mHTT-specific zinc finger protein transcription factors (ZFP-TFs). Viral delivery of these agents, in conjunction with Q175 or Q175-[x] Cre lines, allow for spatially and temporally well-controlled, targeted lowering of mHTT in defined CNS regions throughout the cortico-striatal-thalamocortical circuit and in specific cell types, such as neuron subtypes, astrocytes and oligodendrocytes. Using neurophysiological characterization to evaluate the effect of region-specific mHTT lowering (currently targeting the striatum, GP and STN) will inform about the potential of spatially-restricted mHTT lowering (as in current clinical approaches) to restore circuitry dysfunction in symptomatic animals, using precise neurophysiological characterization. We can then correlate this to behavioral improvement. This will be pivotal in assessing the “where” question within the HTT Lowering MFA to predict the most promising clinical interventions.
The use of mHTT ZFP-TFs will also allow precise assessment of the effect of mHTT lowering in specific cell types on circuitry dysfunction in the Q175 model. And, with the use of the genetic Cre and Cre-ERT2 lines, it will allow us to dissect the genesis of how mHTT expression at a particular locus affects circuit function. In this way we intend to build a detailed understanding of cell-autonomous versus non-cell autonomous neurophysiological dysfunction.
Pharmacological approaches to combat neurodegeneration
Understanding signaling events that precede and precipitate structural neurodegeneration in HD is paramount. At the cellular level HD seems to arise from neurotoxicity involving a prolonged state of cellular dysfunction preceding cell death, due to the intracellular exposure to mHTT and N-terminal HTT fragments containing the expanded polyglutamine domain. Protein aggregation, abnormalities in cellular signaling and trafficking, and the dysregulation of gene expression are implicated, with mutant huntingtin-mediated neurodegeneration the end result of a series of malfunctioning cellular processes inducing neuronal morphological changes that include axonal die-back, dendritic retraction and synaptic loss.
These ultimately neurodegenerative morphological changes are postulated to be the consequence of several processes. These include primary synaptic dysfunction (synaptopathy) resulting in the subsequent pruning of ‘useless’ or damaging connections; disruption of fast axonal transport, including but not limited to lack of trophic factor delivery; and deficient myelination, possibly via a primary oligodendrocyte pathophysiology.
CHDI and collaborators are examining these biological processes and the feasibility and effect of pharmacological intervention, with the aim of validating their role in mutant huntingtin-mediated neuronal dysfunction. There is a rich literature implicating deficits in neurotrophic support and BDNF signaling in HD pathophysiology, and multiple studies suggest that increasing striatal BDNF levels may have therapeutic potential. However, the prospect of restoring BDNF to the striatum is not straightforward, nor is pharmacological small-molecule agonism of the TrkB receptor. Further, recent evidence suggests that defects in TrkB receptor downstream signaling may also be a concern. CHDI-supported studies have recently highlighted an emerging synergy between BDNF signaling deficits, P75NTR signaling upregulation, and the aberrant activation of Rho-GTPase signaling in HD models. This offers an opportunity to intervene in these neurodestructive signaling cascades via a variety of orthogonal and more pharmacologically tractable approaches. Consequently, in addition to investigating the effect of monoclonal TrkB agonizing antibodies on neurophysiological restoration, we are also investigating whether P75NTR antagonism could have beneficial effect. We have also implemented an internally-driven medicinal chemistry effort to generate selective Rho kinase (ROCK) inhibitors, which are known to promote neuronal survival and have shown benefit in HD models. We are also exploring the prospect of restoring fast axonal transport deficits via the inhibition of two kinases, JNK and MLK2/3, the latter also an internal medicinal chemistry program.
Support for ongoing target validation endeavors
We are also assessing the therapeutic potential of other high-interest emerging targets that arise from various sources, such as data from internal or collaborative neurophysiological investigations, or the Unbiased Systems-based Approaches MFA. We also work in collaboration with industry colleagues to evaluate promising pharmacological approaches in preclinical models. For example, our work implicating cyclic nucleotide signaling dysregulation as a proximal deficit contributing to neurophysiological dysfunction in HD led to preclinical validation and the recent clinical trial of Pfizer’s PDE10 inhibitor PF-02545920. In addition to ROCK and MLK inhibition, we are currently evaluating PDE1 inhibition, soluble guanylyl cyclase activation, and M4 positive allosteric modulation for therapeutic potential. Target validation is also being actively pursued around GLT1 activation.
Suggested further reading
Eidelberg D & Surmeier DJ. Brain networks in Huntington disease. J Clin Invest. (2011) 121:484
Plotkin JL et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron. (2014) 83:178
Waldvogel HJ et al. The neuropathology of Huntington’s disease. Curr Top Behav Neurosci. (2015) 22:33
Albin RL & Burke JF. Potential trade-offs in treatment of premanifest Huntington’s disease. Mov Disord. (2015) 30:1319
Mayer IMS & Orth M. Neurophysiology in Huntington’s disease: an update. Neurodegener Dis Manag. (2014) 4:155
Novak MJ et al. Basal ganglia-cortical structural connectivity in Huntington’s disease. Hum Brain Mapp. (2015) 36:1728
Werner CJ et al. Altered resting-state connectivity in Huntington’s disease. Hum Brain Mapp. (2014) 35:2582