
Unbiased Systems-based Approaches MFA
SUMMARY
The biological mechanisms by which the expanded CAG tract of the HTT gene causes the spectrum of HD clinical phenotypes remain unknown, and identifying them will be pivotal for HD therapeutic development. CHDI’s Unbiased Systems-based Approaches MFA strategy has two main directions—to pursue a human genetics approach to correlate genotype to phenotype and to identify genes that modulate the natural history of HD, and to apply a variety of computational approaches to multiple types of unbiased data to reveal the biology that links the huntingtin mutation to the clinical signs and symptoms of HD. Both of these approaches are being undertaken to identify the causal molecular mechanisms of HD and thereby select relevant candidate therapeutic targets.
CHDI is primarily pursuing this goal along two research tracks. First, we are supporting human genetics approaches, initially genome-wide association (GWA) studies, as well as exploring deep sequencing in HDGECs to identify gene variants that modify the natural history of phenotypic manifestations. Second, we are employing computational modeling strategies predicated on interrogating HTT CAG-dependent allelic series—our current focus is on HD mouse models and human stem cells and their derivatives—to probe how omic networks map onto CAG expansion.
RESEARCH VISION
The Unbiased Systems-based Approaches MFA has a unifying theme: To use unbiased data and advanced computational strategies to identify the molecular and circuit-related events that result from the huntingtin mutation. Understanding these disease-modifying biological processes will allow us to identify causal mechanistic pathways and promote targets with therapeutic potential for HD.
Three questions define our priorities with respect to this approach. First, what biological mechanisms underlie the correlations between CAG repeat length and motoric age of onset (as well as other pathogenic phenotypes) in HDGECs? Interfering with these mechanisms may offer the potential to significantly slow disease progression. Second, since mutant huntingtin is broadly expressed in almost all cells, why is HD predominantly a neurodegenerative disease and why is there different susceptibility to dysfunction in various cells and tissues even within the brain? Understanding differential vulnerability may point to ways to block that susceptibility, and identifying compensatory mechanisms that protect unaffected cells may reveal strategies to bolster the vulnerable ones. Third, how do molecular networks respond to the introduction or removal of mutant huntingtin, both in the short and long term? Are the consequences of CAG expansion a relatively linear cascade or is there a starburst of dysfunction? The earliest molecular effects of the CAG expansion are likely to be most proximal to the mutation and to have the broadest causal effect on the disease, and therefore to offer the most promise as disease-modifying therapeutic targets. In juxtaposition, biologically downstream and temporally later changes are more likely to reflect adaptive changes.
The term ‘systems biology’ means different things in different contexts; nevertheless, the endeavor in practice is by nature cyclical. In CHDI’s systems biology program we are interrogating large datasets (initially omics) to compute models to generate explanatory hypotheses. These computational models are then experimentally tested, resulting in new datasets that can be used to improve the validity of our computational models. We are striving for the models to be refined over sufficient cycles so as to be predictive of HD biology. As the cycle turns, the refined computational models are intended to identify targets of interest for therapeutic development.
CHDI’s collaborations in human genetics and computational modeling are envisioned to be enabling to the wider HD research community. CHDI is keen to engage scientists with expertise relevant to the identified human modifier genes so that they can connect these to potential therapeutic mechanisms. The use of unbiased, systems-based approaches in the field of neurodegeneration is still in its infancy. By sharing algorithms, complex datasets and models in freely available formats, CHDI aims to establish a path forward for these approaches for HD, as well as to inform similar approaches in other neurodegenerative diseases. Toward this goal, CHDI and colleagues have developed HDinHD.org, an online portal that links to the GWA datasets and houses an increasing set of large-scale datasets, tools, and computational models created by CHDI and colleagues.
RESEARCH PROGRAM DETAILS
Human genetics
HD is inherited as a dominant allele and invariably occurs in people who carry 40 or more CAG repeats in one of their HTT gene alleles. An inverse relationship exists between CAG repeat length and the age at which HDGECs develop motoric manifestations of the disease, with CAG length determining 55-60% of the variance in age of motoric onset. The remaining variance is contributed by environmental and other genetic factors; identifying these will greatly increase understanding of the disease and help identify avenues to modify clinical outcome.
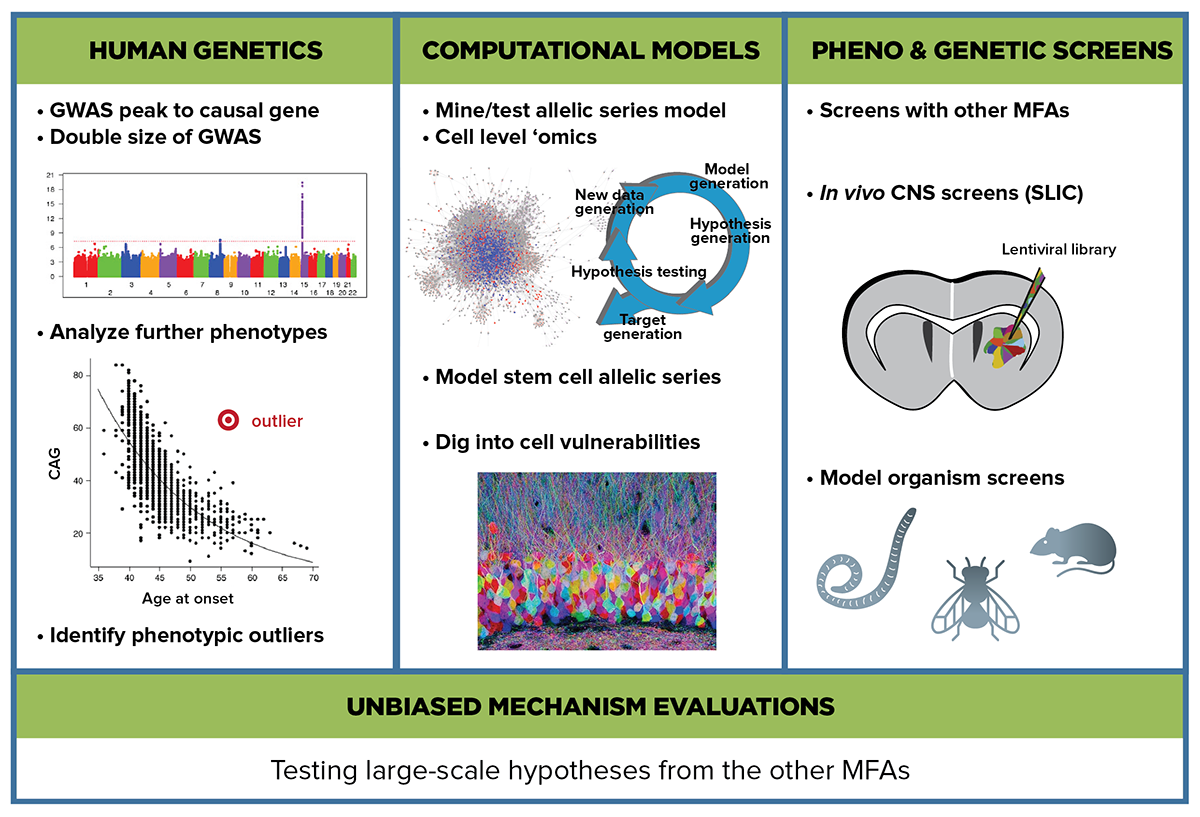
We have been conducting GWA studies in large cohorts of unrelated HDGECs to uncover genetic modifiers of disease phenotypes. By characterizing these human modifiers the resulting candidate targets are by definition biologically relevant, validated in humans, and highly translatable to drug discovery. In recently published GWA studies, CHDI and colleagues identified three loci that contain gene variants either delaying or hastening the motoric age of disease onset. These initial studies suggest that candidate genes within these three loci converge on a fundamental cell process; they are all involved in DNA integrity surveillance and chromatin regulation. This GWA study has now doubled in size and the emerging data indicate a number of additional genetic modifiers will be identified.
While human genetics can identify relevant modifier genes, the biochemical details of how they interact in the context of an expanded HTT CAG allele remain to be defined. This is a high priority area for CHDI. We are now increasing our support for research that examines the biology underlying the modulating effects of these variants in patients, as well as in a variety of animal and cell-based HD models.
 The Unbiased Systems-based Approaches MFA utilizes large unbiased datasets and complex analysis and modeling methods to reconstruct the sequence of molecular events downstream of the huntingtin mutation.
The Unbiased Systems-based Approaches MFA utilizes large unbiased datasets and complex analysis and modeling methods to reconstruct the sequence of molecular events downstream of the huntingtin mutation.
GWA is an unbiased approach that is usually powered to detect common minor alleles that occur in populations alleles that are present in less than 2% of the population by the complementary approach of linkage analyses. Researchers are looking for rare alleles reflecting extreme phenotypes in families or individuals of specific ethnic background, such as Venezuelan families afflicted with HD. These projects also aim to uncover genetic variations that contribute to HD’s age of onset. CHDI is seeking to use the Enroll-HD platform (including expanding beyond European ancestries and geographies/environments) to collect genotype-phenotype relationships in HDGECs, with an increasing focus on identifying those with atypical genotypes and/or phenotypes.
Human genetic studies have historically probed for disease alleles that directly play a role in pathogenesis; searching instead for modifiers of clinical outcome is a novel strategy for Mendelian disorders. The functions of such modifier genes may be contextual in that their biological effect may be revealed in interaction with the HD mutation and have no adverse (or beneficial) effects in its absence. This potential HD gene context-dependent specificity of modifiers may make them especially attractive as targets for therapeutic intervention. In addition, understanding the biological underpinnings of how HD symptoms and clinical outcomes are modulated in different genotypic contexts may also make it easier to design and interpret interventional trials and to analyze other genetic studies.
Computational modeling
The interplay between molecular networks and cellular circuitry in the brain on the backdrop of the degeneration that characterizes HD makes for a degree of complexity that undoubtedly exceeds what a single human brain can comprehend. We aim to harness the power of modern computing algorithms to enhance HD researchers’ ability to sort through enormous quantities of data and to come up with novel hypotheses about the cascade of physiological and molecular events that underlie the disease.
Currently, the data that these computational strategies will use come from CHDI-supported investigations of an in vivo mouse allelic series—we have explored multiple HD mouse models that carry increasing HTT CAG repeat lengths to determine how phenotype plus genomic, transcriptomic, proteomic, and other omic networks map to the CAG expansion. Recently CHDI and colleagues reported in mice that the striatum—one of the brain regions most vulnerable to HD-related neuronal pathology—carries a transcriptional signature dependent on CAG length. Also of interest, CHDI and collaborators have discovered a CAG-dependent signature in cerebellum, a tissue widely thought not to be greatly affected in HD. Contrasting the networks across brain regions/cell types with different vulnerabilities will likely shed light on the cellular responses to mutant huntingtin, and some may have therapeutic promise.
This framework will allow researchers to address several key unknowns. First, we can explore whether CAG repeat length corresponds linearly to time of onset, or whether especially long (or short) CAG lengths have specific phenotypic effects. This will require transcriptomic analyses of old and young animals at both extremes of CAG repeat length. As mentioned above, a critical question is whether the pathophysiological cascade downstream of the huntingtin mutation perturbs a limited number of linearly-linked mechanisms or is a starburst of many concurrently dysregulated mechanisms. Preliminary work points to the former, but answering this question will require expanding our investment in omics studies to collect data on multiple levels, particularly, in the near term, with respect to the epigenome.
We can also ask whether human cells—for example primary cells, pluripotential stem cells and derivatives—will be a valuable computational modeling platform for HD and, more broadly, whether CHDI should be prioritizing tissue- or cell-level measurements. Most omics studies in HDGECs and animal models have been conducted using dissected tissue (which is limited in supply), but it is now technologically possible with cell-type and single-cell profiling approaches to explore these questions with potentially both more power and more refinement. It remains unclear how much the loss of connectivity to other neurons and glial cells lowers the value of cell-based assays when utilizing computational models for biological and therapeutic insights. We are currently conducting allelic series experiments in both human and mouse cells to investigate this question and are exploring emerging new areas.
Overall, CHDI is still learning how our computational models that are designed to make connections between multiple biological levels in an unbiased manner (for example mRNA to protein to cellular circuitry) will integrate the many biological layers that we are probing into a single knowledge framework. Although much of the preceding efforts come from oncology and inflammation, our computational algorithms will rely primarily on unbiased neurobiological datasets that CHDI and colleagues generate to reconstruct molecular networks; we are optimistic this will reveal new insights relevant to pathogenesis and potential therapeutic interventions. CHDI is very interested in engaging computational modelers with diverse expertise to apply their approaches to the HD datasets made available at HDinHD.org.
Other approaches
CHDI is beginning to explore large-scale unbiased phenotypic screens. Successes from these unbiased screens will identify previously-unsuspected genes modulating biological mechanisms likely involved in HD. We are specifically focused on in vivo phenotypic screening, currently using fruit flies and mice. Additionally, we are exploring a strategy for unbiased mechanism evaluation where we will investigate specific molecular mechanisms in parallel across a number of HD models to elucidate relevant global changes. Mechanisms currently under consideration include chromatin structure and genome-wide mis-splicing.
Suggested further reading
Alexandrov V et al. Large-scale phenome analysis defines a behavioral signature for Huntington’s disease genotype in mice. Nat Biotechnol. (2016) 34:838
Langfelder P et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. (2016) 19:623
Bragg RM et al. Motivational, proteostatic and transcriptional deficits precede synapse loss, gliosis and neurodegeneration in the B6.Htt(Q111/+) model of Huntington’s disease. Sci Rep. (2017) 7:41570
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) 162:516
Ament SA et al. High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet. (2017) 26:913
Parmentier F et al. Pathways to decoding the clinical potential of stress response FOXO-interaction networks for Huntington’s disease: of gene prioritization and context dependence. Front Aging Neurosci. (2013) 5:22
Shema R et al. Synthetic lethal screening in the mammalian central nervous system identifies Gpx6 as a modulator of Huntington’s disease. Proc Natl Acad Sci U S A. (2015) 112:268