
HTT Lowering MFA
SUMMARY
The HTT Lowering MFA aims to develop therapeutic strategies that will substantially improve the lives of HD-affected individuals. One guiding principle is to pursue strategies that are most proximal to the cause of the disease—the mutant HTT gene. CHDI will focus on approaches to edit the mutant gene, lower levels of mHTT mRNA, or decrease mHTT protein, either directly or by intervening in pathways and modulating targets that affect huntingtin mRNA and protein levels—this will define what will comprise a therapeutic. For each therapeutic modality we will empirically address when in disease course is optimal to treat, how much lowering of mutant huntingtin is sufficient but safe and where (systemic, whole brain, brain subregions, specific cell types) nucleic acid or virally-delivered gene therapies need to be delivered to maximize beneficial clinical outcomes. To improve the mode of drug delivery we are also working to identify small-molecule modulators of huntingtin mRNA and protein levels. Also critical to our strategy is developing sensitive and robust pharmacodynamic (PD) biomarkers that can track molecular and non-molecular events that correlate with the activity of HTT-lowering agents in HDGECs.
RESEARCH VISION
Since HD is caused by a single gene mutation, some of the most promising therapeutic targets include the expanded mutant HTT gene, mRNA, and translated protein. CHDI and colleagues, as well as many others in the wider HD research field, are actively working to better elucidate the complex huntingtin life cycle by studying huntingtin structure-function (see HTT Structure & Function MFA synopsis), with the ultimate goal of identifying the most promising pharmacological targets for therapeutic intervention.
The multifaceted huntingtin life cycle offers a multitude of possibilities (Figure 1). Emerging gene editing technologies now allow us to pursue targeting of the causative mHTT gene itself. The mHTT mRNA adopts a secondary structure that may contribute directly to pathogenesis—for instance by promoting aberrant or mis-splicing of exons, formation of RNA foci, or sequestration of critical RNA splicing factors that affect other genes—in addition to its translation to mHTT protein, so targeting mHTT mRNA transcription or promoting mRNA degradation is a high priority.
Mutant HTT protein species—full length, fragments and post-translationally modified forms—aggregate and sequester other critical proteins in the cell but further insight is needed to resolve the complexities of the production, misfolding and aberrant protein-protein interactions. We aim to develop better tools and assays to define aggregated forms of mHTT for tracking during interventions to prevent production or remove these species.
The HTT Lowering MFA has prioritized the following principles for the development of HTT-lowering therapeutics:
Proximity to the mutation: Intervention as close as possible to the genetic insult.
Selectivity for mutant huntingtin: Specifically target the mutant huntingtin gene, RNA or protein; pan-huntingtin approaches to lower both mutant and non-expanded will be pursued but with lower priority.
Early treatment: Intervene as early as possible to achieve maximal therapeutic benefit while ensuring patient safety.
Broad huntingtin-lowering throughout the CNS: Wide distribution in the CNS to as many cell types as possible; also in the periphery if feasible.
Strategic breadth: Removing the genetic insult or clearing newly-synthesized mHTT mRNA may not be sufficient to provide meaningful benefit to patients with manifest disease, so strategies to identify and clear long-lived mHTT protein species will also be pursued.
RESEARCH PROGRAM DETAILS
HTT-lowering therapies
Gene-level interventions Gene-editing technologies could be the most direct therapeutic approach, with the potential to reduce the CAG repeat length, interfere with its transcriptional machinery, or delete the gene selectively by targeting either single nucleotide polymorphisms in the mutant allele or the CAG expansion. Specific technologies—including zinc finger nucleases, transcription activator-like effector nucleases (TALENs) and CRISPR/Cas9—have already been applied to make important proof-of-concept modifications to the HTT gene in cell and animal model systems. However, the development of these tools into applied therapeutics is still at an early discovery phase for HD and questions remain regarding the best strategy to edit mHTT to attain maximal editing efficiency with minimal off-target effects. Over the next three years CHDI will conduct proof-of-concept studies and work with academic and industry collaborators to improve the selectivity and efficiency of gene editing for HTT, as well as the best way to deliver these gene therapy agents to the brain. CHDI is also actively engaged in identifying companies with a therapeutic interest in developing gene-editing approaches for HD.
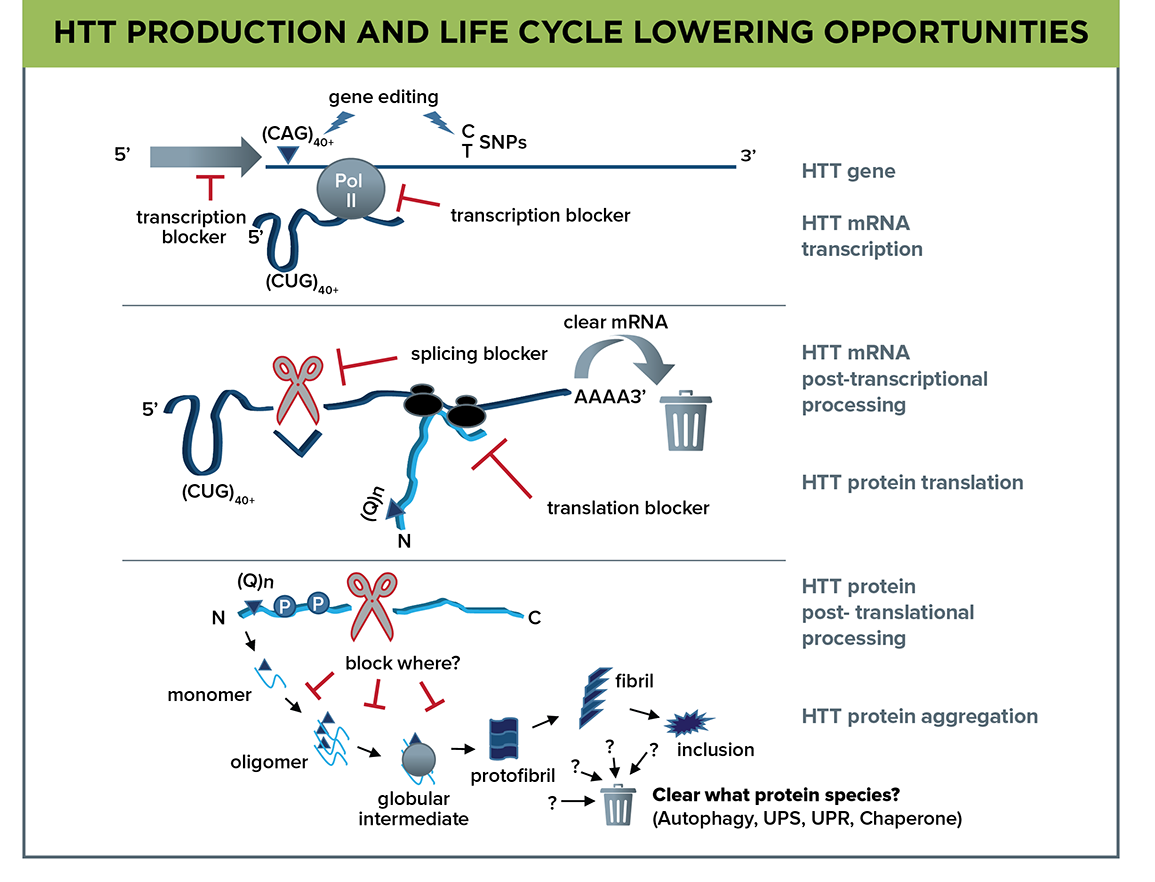
Figure 1. The complexity of the huntingtin life cycle—gene to mRNA to protein—offers a myriad of intervention points; defining which will be the most therapeutically promising is central to the HTT Lowering MFA. The mHTT gene is a target for gene editing at either the CAG expansion or SNPs, and transcription blockers can act on mRNA initiation at the gene promoter or at Pol II transcription initiation or elongation. HTT mRNA is a target for clearance, blockers of mRNA splicing, or inhibition of protein translation. mHTT protein is a complicated target due to the array of species formed by proteolytic cleavage, post-translational modifications, and misfolding leading to oligomers, fibrils, and inclusion bodies.
RNA-level interventions Blocking transcription – CHDI is investigating the therapeutic potential of interfering with HTT transcription via two different strategies. First, we are supporting investigations that use zinc finger protein transcription factors (ZFP-TFs) to repress the transcription of the mHTT allele, and investigating whether inhibiting the expression of mHTT of various CAG repeat lengths can correct disease phenotypes in our mouse allelic series (see Unbiased Systems-based Approaches MFA synopsis). We will also use ZFP-TFs to probe the degree to which the normal HTT allele can be repressed, which will give us a better understanding of the potential loss of normal huntingtin function this could entail. ZFP-TFs will be delivered to model systems using AAV vectors but we will also generate mouse models that allow their conditional expression. This work may lead to identification of safety markers that could track adverse events associated with total HTT levels dropping below a critical threshold.
Second, inhibiting the Supt4H gene—which promotes RNA polymerase II-mediated transcription through regions of DNA containing trinucleotide repeats—could also be a mHTT transcription-blocking strategy. CHDI will explore genetic and small-molecule inhibitors, when available, to block Supt4H activity and measure the effects of selectively lowering mHTT mRNA transcription in cell and animal models.
Promoting mRNA degradation – Multiple technologies to lower mHTT mRNA are also under investigation, and we are focusing specifically on several fundamental biological mechanisms; RNA interference through RISC/Dicer, and antisense oligonucleotides that work either via RNAse H nuclease activity to degrade mRNA or through blocking translation. Each of these are at different stages of development, with the most advanced being the Ionis Pharmaceuticals HTT ASO currently in clinical trial. Other biotechnology companies are developing virally-delivered RNAi gene therapy approaches targeting total HTT mRNA. CHDI is also working with academic colleagues to develop new chemically-modified siRNAs to improve potency, biodistribution, and durability in vivo. Working directly with or enabling industry colleagues, CHDI is advancing RNA interference-based therapies towards the clinic, as well as investigating the best ways to deliver these agents to maximize distribution to the brain and enabling the use of PD/proximal biomarkers for HTT lowering.
Small-molecule modulators of HTT levels Due to their better systemic distribution and ease of administration, a high priority is to identify orally-available small-molecule drugs that modulate huntingtin protein or mRNA levels. For economic reasons such drugs would also be more accessible to all HD patients, including those in developing countries. CHDI is working with several CROs and biotech/pharma companies to identify HTT-lowering small molecules in unbiased phenotypic screens that employ HD patient-derived cells, including induced pluripotent stem cells and human embryonic stem cells in undifferentiated and neuronally-differentiated states. The goal is to identify both compounds and mechanisms that regulate mutant huntingtin levels and advance those with therapeutic potential.
HTT protein clearance In collaboration with the HTT Structure/Function MFA, studies have been initiated to investigate the misfolding of mHTT and how it forms oligomers, fibrils, and insoluble inclusion bodies, as well as to define aberrant protein-protein interactions within cells that are driven by mHTT. Importantly, we are developing new tools and assays to better define the misfolded mHTT conformers, and these will be used in preclinical interventional studies aiming to lower mHTT. The overall goal is to precisely define the species of mHTT protein(s) that must be cleared to improve phenotypes in our model systems. In parallel, CHDI will continue to investigate ways to modulate proteostatic and protein clearance pathways involved in processing mHTT, prioritizing autophagy but also exploring the ubiquitin-proteasome, unfolded protein response, and chaperone response pathways in a CNS context. Towards this goal, we are exploring a variety of reporter systems to identify the main mechanisms implicated in modulating mHTT expression in the CNS.
CHDI is also investigating HTT-specific therapeutic antibodies that can lower mHTT; such agents are increasingly being developed for other neurodegenerative disorders, particularly Alzheimer’s disease, and recent data is showing promise. The HTT epitopes and the conformers to which each antibody binds will be defined, and their ability to lower mHTT tested in cell and animal models.
Delivery technologies
Due to their poor blood brain barrier (BBB) penetration, current nucleic acid therapeutic approaches targeting HTT DNA or RNA must be administered directly to the CNS, either into the CSF (intrathecally or intracerebrovascularly) or the brain (intraparenchymal infusion). This has resulted in only limited CNS distribution. CHDI is interested in technologies to improve the transit of such large agents across the BBB to efficiently deliver them to the brain and we will adopt innovative approaches as they are developed. Areas of active investigation showing promise include the development of nanocarriers, exosomes, and liposomes.
CHDI and collaborators are also exploring adeno-associated virus (AAV), and alternatives such as paramyxoviruses, that can be genetically engineered to deliver therapeutic DNA or RNA into neurons and other brain cells after central delivery. AAV-based gene therapies for several neurological disorders are in clinical trials, and some serotypes have been approved for clinical use by the FDA and EMA. One advantage of AAV delivery is long-term expression and efficacy as demonstrated in both animal and human studies; AAV-delivered agents can be expressed for 10 years or more. Once in the brain, AAVs can be transported along axons to transduce neighboring neurons, so an AAV-delivered therapy might be passed to some extent through affected brain circuitry. Investigations are underway on how different AAV serotypes administered differently affect the distribution of virus and its cargo, as well as how each targets mutant huntingtin gene, mRNA or protein in the brain. CHDI is enabling these strategies by making large HD animal models available for such studies, as well as developing new primate and farm animal models.
Novel AAV serotypes and paramyxoviruses, engineered to target specific cell-surface receptors, are part of our early exploratory work to investigate whether these vectors can cross the BBB to deliver huntingtin-lowering therapeutics to specific neuronal or glial cell types. To better understand and develop this delivery methodology, CHDI has established a gene therapy key opinion leader panel to advise on research strategies.
HTT-lowering biomarkers
HTT-lowering PD biomarkers are molecular or non-molecular indicators that lowering agents are engaging the target of interest and having a pharmacological effect in HDGECs. Ideally, such PD biomarkers will be used to establish a link between the pharmacological agent, the target effect in a region of interest, and the biological response. We are currently investigating molecular, neuroimaging, and physiological biomarkers in disease models that have potential to translate to humans. These efforts are being guided by a CHDI HTT-lowering biomarker task force.
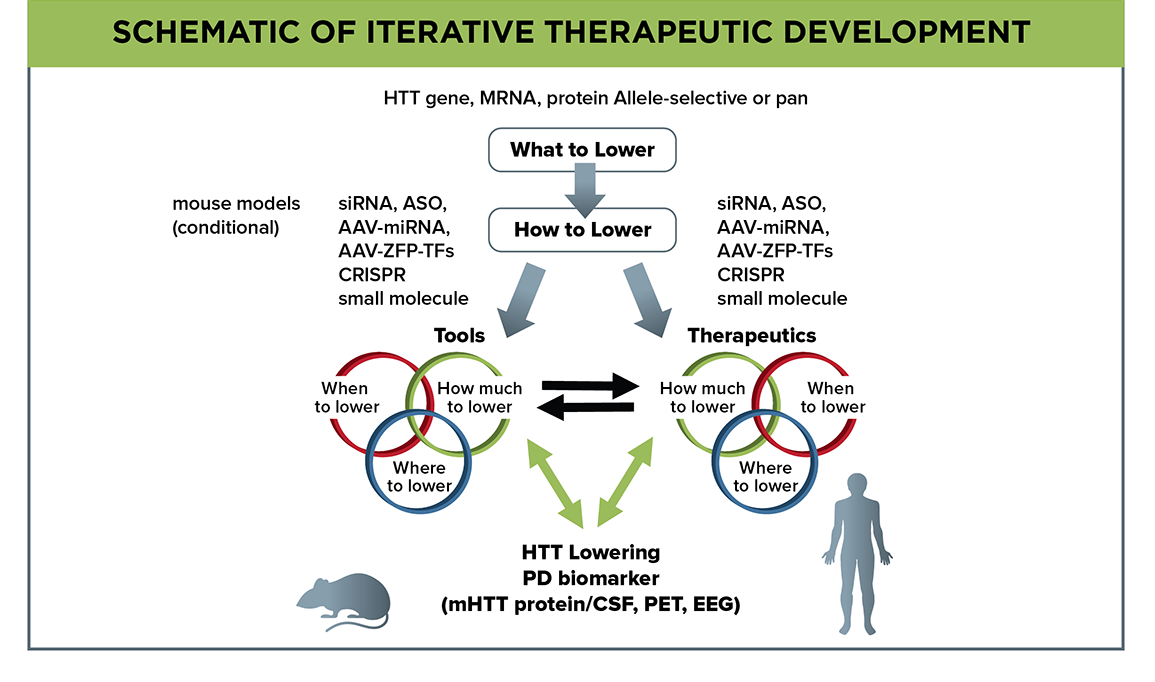
Figure 2. Flow schematic depicting empirical investigations to address what, how, when, where, and how much to lower mutant huntingtin. Model-system investigation of huntingtin-lowering pharmacodynamic biomarkers will aid their development for use in clinical trials.
CHDI has focused on the measurement of mHTT protein itself as a molecular PD biomarker. With colleagues we have developed multiple platforms—including Meso Scale Discovery (MSD), Singulex Erenna, and time-resolved fluorescence resonance energy transfer (TR-FRET)—to assay mHTT in various biosample types. Notably, we can now measure mHTT in the femtomolar range in human CSF, and this can now be used to assess the action of central huntingtin-lowering agents in clinical trial. We will also apply advanced proteomics techniques, including discovery and targeted mass-spectrometry proteomics, to identify other potential molecular PD biomarkers. Targeted proteomics will look for individual proteins in human and animal model CSF that are altered during disease progression since such proteins may respond to HTT-lowering agents. Discovery proteomics may define protein signatures that are altered due to HTT lowering.
Neuroimaging biomarkers are essential tools, particularly those with utility in positron emission tomography (PET). Several PET tracer studies have already identified markers (including phosphodiesterase 10A) that are dysregulated in HD patients, and we are investigating how these markers respond to HTT-lowering agents in model systems. Importantly, CHDI and collaborators have developed several novel small-molecule binders of aggregated mHTT that, when radiolabeled, can function as PET tracers, and these are now being evaluated in animal models and are on track to be tested in humans in 2017.
We are also investigating quantitative EEG (qEEG) and MEG—noninvasive measures of neuronal brain activity—as a physiological PD biomarker of HTT lowering that has already been applied successfully to show changes in premanifest and manifest HD patients, as well as multiple mouse models. We are now determining whether qEEG and MEG signatures in HD models are altered by HTT-lowering agents before developing for humans.
What, how, when, where, and how much to lower mutant huntingtin?
Despite several HTT-lowering therapies already being in clinical development, some major questions remain (Figure 2). What the best target is within the huntingtin lifecycle remains undefined, and may be at the gene, mRNA or protein level. Our priority is to emphasize developing strategies that target each selectively, while leaving the normal HTT allele (and its expression) spared. How we will target the huntingtin lifecycle components will inform us on additional parameters that must be empirically addressed; this includes when in the course of disease a specific modality must be administered to be most effective. Will a therapeutic still offer benefit if given to patients with advanced disease, or will it only reduce clinical signs and symptoms when administered before these emerge, or at some other prodromal stage? Since these questions will be initially addressed in animal models, we need to determine the correlation between disease progression timelines in animal models and humans.
We also need to determine empirically where HTT-lowering therapies are required—will they be required throughout the entire body, the entire brain, or can we see meaningful therapeutic benefit when covering only subregions of the brain? Will it be sufficient to lower mutant huntingtin in neurons, and if so which neurons, or do we need to lower in other cell types? Working with the Neuronal Dysfunction MFA, we are addressing where the most effective site is within the cortico-basal ganglia-thalamo-cortical circuit to deliver a therapeutic for maximum benefit to circuit restoration and symptom improvement. Within these regions, emphasis has been placed on two separate evaluations of mutant huntingtin lowering, targeting either predominantly neuronal populations or exclusively within astrocytes and oligodendrocytes. Ideally, the goal is therapeutic approaches that target all cell types simultaneously and effectively throughout the brain; we will use conditional mouse models and broadly-distributed HTT-lowering agents to investigate, including novel AAV serotypes that can be administered peripherally and result in broad coverage of neurons and astrocytes in the mouse brain.
Finally, we need to assess how much each therapeutic modality must lower mutant huntingtin to achieve maximal therapeutic benefit. How much lowering of mutant huntingtin and, importantly, of total huntingtin is sufficient but also safe? As huntingtin has normal functions, strategies that especially target total huntingtin have a potential toxicity profile. Working with the HTT Structure/Function MFA, we are using genetic and viral-delivery methods in rodents to address the potential safety issues associated with loss of huntingtin function in the adult CNS. Our focus will be on identifying markers associated with loss of normal huntingtin function.
These empirical questions of where, when, and how much to lower mutant huntingtin will be evaluated in model systems for each specific modality; these data will be crucial to clinical trial prioritization and design.
Suggested further reading
Aronin N & DiFiglia M. Huntingtin-lowering strategies in Huntington’s disease: antisense oligonucleotides, small RNAs, and gene editing. Mov Disord. (2014) 29:1455
Munoz-Sanjuan I & Bates GP. The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest. (2011) 121:476
Mestre TA & Sampaio C. Huntington disease: Linking pathogenesis to the development of experimental therapeutics. Curr Neurol Neurosci Rep. (2017) 17:18
Rubinsztein DC & Orr HT. Diminishing return for mechanistic therapeutics with neurodegenerative disease duration? There may be a point in the course of a neurodegenerative condition where therapeutics targeting disease-causing mechanisms are futile. Bioessays. (2016) 38:977
Wild EJ et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J Clin Invest. (2015) 125:1979
Carroll JB et al. Treating the whole body in Huntington’s disease. Lancet Neurol. (2015) 11:1135
Kordasiewicz HB et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. (2012) 74:1031
Keiser MS et al. Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from Huntington’s disease and spinocerebellar ataxia. Hum Mol Genet. (2016) 25(R1):R53
Saudou F & Humbert S. The biology of huntingtin. Neuron. (2016) 89:910
Byrne LM & Wild EJ. Cerebrospinal fluid biomarkers for Huntington’s disease. J Huntingtons Dis. (2016) 5:1